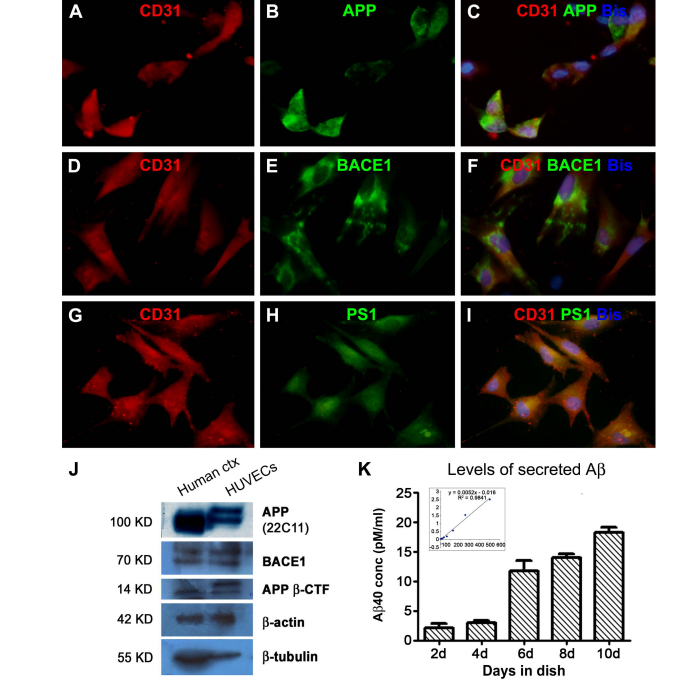
 Cholesterol Potentiates β-Amyloid Genesis in Cultured Human Umbilical Vein Endothelial Cells Guozhi Huang1,2, Zhiqin Xue3, Xia Hu1, Lily Wan1, Jianming Li4, Yan Cai1, Peter R. Patrylo5, Xuegang Luo1, Aihua Pan1, Xiao-Xin Yan1,6* 1 Department of Anatomy and Neurobiology, Central South University Xiangya Medical School, Changsha, China 2 Department of Anatomy & Embryology, Loudi Vocational and Technical College, Loudi, China 3 Department of Anatomy, Xinjiang Medical University, Urumqi, China 4 Neuroscience Research Center, Changsha Medical University, Changsha, China 5 Departments of Physiology, Anatomy and the Center for Integrated Research in Cognitive and Neural Sciences, Southern Illinois University Carbondale, Carbondale, USA 6 Key Laboratory of Hunan Province in Neurodegenerative Disorders, Changsha, China Received 20 April 2016; accepted 3 June 2016; published 6 June 2016 Copyright © 2016 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Cerebral Amyloid Angiopathy (CAA) occurs commonly among the elderly and almost invariably in patients with Alzheimer’s Disease (AD). The β-amyloid peptides (Aβ) are produced via the amyloidogenic processing of β-Amyloid Precursor Protein (APP) by β-secretase-1 (BACE1) and γ- secretase. Vascular endothelial cells are lately shown to possess the molecular machinery of Aβ production, which might participate in the development of CAA. Hypercholesterolemia is considered a risk factor for AD, whereas less is known if cholesterol may modulate endothelial Aβ production. In the present study we verified the amyloidogenic capability of Human Umbilical Vein Endothelial Cells (HUVECs) in vitro and explored the effect of cholesterol exposure on their amyloidogenic potential. Cholesterol treatments at 12.5 and 25 mg/dL significantly elevated APP, BACE1 and APP β-CTF protein levels and β-site APP cleavage activity in cell lysates, and Aβ40 levels in culture medium. However, coincubation with cholesterol at 50 and 100 mg/dL attenuated the viability of the cultured cells and diminished their amyloidogenic capability. These findings suggest that high cholesterol exposure is stressful to vascular endothelial cells, and at a certain dosage range can promote an amyloidogenic response in these cells. * Corresponding author. G. Z. Huang et al. 24 Keywords Aging, Alzheimer’s Disease, Cholesterol, Neurodegeneration, Vascular Dementia 1. Introduction Cerebral amyloidosis is a pathological condition caused by the deposition of the β-amyloid peptides (Aβ) in the extracellular matrix and vascular wall in the central nervous system, referred to as parenchymal amyloid plaques and Cerebral Amyloid Angiopathy (CAA), respectively [1] [2]. Amyloid plaques and CAA are among the definitive neuropathological lesions of dementia of the Alzheimer’s Disease (AD) and vascular types [3]-[5], although both types of amyloidosis can be seen in the brain of non-demented elderly [6]-[13]. Along with Aβ deposits, parenchymal and vascular amyloid pathology may coexist with degenerative and reactive cellular changes, including neuronal and vascular cell loss, neuritic dystrophy and glial activation [14]-[19]. Neurons appear to be the major origin of soluble and insoluble Aβ products in the brain given that they are enriched with the amyloidogenic biochemical machinery including β-Amyloid Precursor Protein (APP), β-secretatse-1 (BACE1) and γ-secretase [20]-[22]. In particular, elevated BACE1 protein and enzymatic activity are reported in the human brains with amyloid neuropathology [22]-[30]. Specifically, enhanced BACE1 expression is identified microscopically in dystrophic axons around neuritic and angiopathic Aβ deposition [18] [31]-[34]. Vascular endothelial cells are initially shown to express the full set of amyloidogenic machinery in vitro, which has been lately demonstrated in vivo including in the human brain with CAA [35]-[42]. It is suggested that Aβ overproduction by vascular endothelial cells may be involved in the early stages of the development of CAA [40]-[42]. Therefore, it becomes important to identify circulatory factors that might potentiate endothelial amyloidogenesis. Hypercholesterolemia has been considered a risk factor for AD and vascular dementia for a long time [43]-[53]. However, it remains less clear if cholesterol may directly influence the amyloidogenic activity in vascular endothelial cells [54]. In the present study, we used Human Umbilical Vein Endothelial Cells (HUVECs) as a model to explore a modulatory effect of cholesterol on endothelial amyloidogenic protein expression and Aβ secretion. Cultured HUVECs exhibited a biphasic response to cholesterol treatments, with low dose exposure potentiated Aβ genesis while high dose exposure resulted in cell death and diminished Aβ production. 2. Materials and Methods 2.1. Cell Culture and Cholesterol Treatment HUVECs were obtained commercially (Catalog #C0035C, Life Technologies Corporation). Cell supplies were re-suspended with DMEM/F12 containing 10% fetal bovine serum, to a beginning density approximately 5 × 106 cells/mL. Cells were cultured at 37˚C in a humidified atmosphere containing 95% air and 5% CO2, in six-well Corning-Costar plates (Catalog #CLS3516; Life Technologies Corporation). Each well was loaded with 2 ml of medium and a glass coverslip coated with polylysine, with the medium changed twice a week. Initial experiments were carried out to verify the cultured cells and optimize the biochemical assays. Cholesterol treatments to HUVECs were initiated on day 4 of culture for each round of experiments for duration of 48 hours. A total of 3 experiments were carried out separately in different times. For each experiment, media containing 0, 12.5, 25, 50 and 100 mg/dL of cholesterol (Catalog #C8667, Sigma-Aldrich, St. Louis, MO, USA) were added in duplicate into wells loaded with the same amount of cells from the same original batch of HUVECs expansion. Media were collected following 48 hours of culture to assess Aβ40 concentrations, with the corresponding groups of cultured cells harvested for morphological study and assessment of related proteins and APP β-cleavage activity. 2.2. Immunocytochemistry and Microscopic Examination HUVECs grown on coverslippers were fixed in situ with cold 4% paraformaldehyde for 20 minutes and stored frozen until immunofluorescent processing. Coverslippers were thawed to room temperature prior to staining, and incubated in PBS containing 5% donkey serum for 30 minutes, followed by reaction with a pair of primary G. Z. Huang et al. 25 antibodies raised in different species overnight at 4˚C, including 1) mouse anti-CD31 (endothelial marker) [41] and rabbit anti-APP C-terminal (CT20); 2) mouse anti-CD31 and rabbit anti-BACE1 [20] [32]; 3) mouse antiCD31 and rabbit anti-presilinin-1 (PS1) N-terminal (ab14) [41]. Following several rinses with PBS, the coverslippers were further incubated at room temperature for a 2 hour with Alexa Fluor® 488 donkey anti-rabbit and Alexa Fluor® 594 conjugated donkey anti-mouse IgGs (1:200, Invitrogen, Carlsbad, CA, USA). Coverslippers were finally counter-stained with bisbenzimide (Hoechst 33,342, 1:50,000, Catalog #B2261, Sigma-Aldrich), washed thoroughly, and mounted with anti-fading medium. Immunofluorescence was examined on an Olympus fluorescent BX53 microscope equipped with a digital imaging system (CellSens Standard, Olympus Corporation, Japan). 2.3. Western Blot HUVECs pellets (×4, w⁄v) were homogenized by sonication in T-PER extraction buffer (Pierce, Rockford, IL, USA) containing protease inhibitors (Roche, Indianapolis, IN, USA). The lysates were centrifuged at 15,000 g, with supernatants collected and protein concentrations determined by DC protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Human cortical lysates available from a previous study (Xue et al., 2015) were used as an assay control to cross-validate the protein profile of the HUVECs relative to brain tissue. Fifty microgram protein was run on each lane in 10% or 18% (for APP β-CTF) SDS-polyacrylamide electrophoresis gels. Separated proteins were electrotransferred to Trans-Blot pure nitrocellulose membranes (Bio-Rad Lab.), which were immunoblotted with antibodies to APP (22C11), APP β-CTF (AHP538), BACE1, PS1-NTF (ab14), β-tubulin-III, β-actin at pre-optimized working dilutions with agitation for 12 hours. The membranes were further reacted with horseradish peroxidase (HRP)-conjugated goat anti-rabbit or anti-mouse IgGs (1:20,000; Bio-Rad Laboratories), treated with the ECL Plus Western Blotting Detection Kit, and exposed against Kodak X-OMAT Blue autoradiography films that were developed in a darkroom subsequently. 2.4. APP β-Site Cleavage (BACE1) Activity Assay APP β-site cleavage activity (representing BACE1 enzymatic activity) in HUVECs was measured in duplicate in 96-well transparent flat-bottomed plates. Samples were homogenized on ice as described above for Western blot, and assayed for enzymatic activity using a commercial kit (Calbiochem, La Jolla, CA, USA, Catalog #565785) according to manufacturer’s instructions. The fluorescent signal was captured in a Bio-Rad microplate reader (PR 3100 TSC). 2.5. Enzyme-Linked Immunosorbent Assay for Aβ in Culture Medium Aβ levels in cell culture media were obtained to estimate the amount of secreted peptides following cultivation for different time periods or treatments of cholesterol at different concentrations. Media were assayed in duplicate by solid sandwich enzyme-linked immunosorbent assay (ELISA) to assess Aβ40 levels using commercial kits (Novex® KHB3482 for Aβ40) following the manufacturer’s instruction (Life Technologies Corporation). Signals were captured in the Bio-Rad microplate reader, with the Aβ concentrations (pM/ml) calculated according to the standard curve generated from the readouts of the synthesized peptides supplied along with the kit. 2.6. MTT Cell Viability Assay The viability of HUVECs was assayed at the end of cholesterol treatment experiments using a commercial fluogenic 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) kit following the manufacturer’s instruction (Abnova, Catalog # KA1606, Taipei City, Taiwan). Fluorescent signal was measured in the Bio-Rad microplate reader, with cell viability calculated for comparing culture groups. 2.7. Quantification, Statistical Analysis and Figure Preparation Densitometric (Western blot, digitalized using NIH-Image J) and numeric data (light absorbent signals of APP β-site cleavage enzymatic activity, Aβ ELISA and MTT fluogenic activity) were normalized to either the internal standard or the control group. Means were calculated for comparing groups, and analyzed statistically using one-way ANOVA with Bonferroni’s multiple comparison tests (Prism GraphPad, San Diego, CA). The significant level of difference was set at P < 0.05. Figures were assembled with Photoshop 7.0, with brightness/ G. Z. Huang et al. 26 contract adjusted as needed. 3. Results 3.1. HUVECs Expressed Amyloidogenic Proteins and Secreted β-Amyloid Peptide Cultured cells from the commercial HUVECs supply were examined immunocytochemically to verify their cellular profile. Cells grown 4 - 10 days in vitro were mostly polygonal and sometimes fusiform in shape, had short somal protrusions and processes. All these cells expressed CD31, a marker of the vascular endothelia (Figure 1(A), Figure 1(D) and Figure 1(G)). In double immunofluorescence, virtually all CD31 expressing cells also co-labeled with antibodies to APP (Figures 1(A)-(C)), BACE1 (Figures 1(D)-(F)) [20] [32] and PS1-NTF (Figures 1(G)-(I)) [41]. To check the expression of amyloidogenic proteins biochemically, HUVECs lysates were blotted in parallel with human cortical lysates (available from our recent study). In comparison, HUVECs lysates contained less abundant holo-APP relative to brain samples, with the blotted protein bands migrated at a slightly heavier molecular weight position in the former (Figure 1(J)). Mature BACE1 (~70 Kd) and APP β-CTF (~14 Kd) were present in cortical and HUVECs lysates at similar migration position (Figure 1(J)). The signal of β-actin was comparable between the two assayed samples, whereas that of the neuronal protein β-tubulin was much more intense in the cortical relative to endothelial lysates (Figure 1(J)). To check Aβ secretion by HUVECs, the amounts of Aβ40 (considered to be most abundant Aβ species in CAA) in culture media were assayed with an ELISA kit. In several preliminary trials with media collected following 3 days of culture, Aβ40 levels were found to be around or slightly above the detecting threshold of kit. Subsequently, a batch of culture was carried out such that inpidual wells were maintained to different time points without change/resupply of culture medium. Thus, media from the wells with cells cultured for 2, 4, 6, 8, and 10 days were assayed for the levels of Aβ40. The amount of Aβ40 in the medium showed a trend of accumulation in the groups cultured from 2 - 10 days (Figure 1(K)). 3.2. Cholesterol Exposure Affected the Viability of Cultured HUVECs Pilot studies were carried out with cholesterol at titered concentrations from 200 down to 6.25 to mg/dL and duration of culture time from 24 - 48 hours, which indicated that the 6.25 mg/dL treatment had no noticeable effect relative to the 0 mg/dL group, whereas 200 mg/dL treatment caused massive cell death. Therefore, formal experiments (3 separate sets) were designed to determine the effect of 48 hour cholesterol coincubation at 0, 12.5, 25, 50 and 100 mg/dL concentrations, respectively. At the end of the treatments, we assessed cell morphology and the amount of cells microscopically as well as their viability using the MTT assay. Cell pallet lysates and culture media were assayed biochemically to quantify amyloidogenic protein and Aβ secretion. On microscopic examination with phase-contrast imaging, cells in the groups cultured with cholesterol at 0, 12.5 and 25 mg/dL were well attached to the coverslippers and showed healthy-looking morphology and comparable density (Figures 2(A)-(C) and Figures 2(A’)-(C’)). In contrast, cells in the 50 and 100 mg/dL groups appeared reduced or largely lost, with small-sized round profiles (likely cellular remains or debris) seen over microscopic field and only a few fusiform cells distinguishable (Figure 2(D) and Figure 2(D’), Figure 2(E) and Figure 2(E’)). In parallel, cell viability was affected by cholesterol coincubation. Thus, relative to control group (0 mg/dL), the MTT scores of the cholesterol treatment groups showed a dose-dependent reduction (P < 0.0001, F = 32.6, df = 4, 10, one-way ANOVA). Posthoc tests (Bonferroni’s Multiple Comparison Test) indicated a significant difference between the 0 mg/dL and all other dosage groups; between the 12.5 relative to the 50 and 100 mg/dL groups, and between the 25 relative to the 50 and 100 mg/dL groups (Figure 2(F)). 3.3. Cholesterol Exposure Affected the Amyloidogenic Capability of Cultured HUVECs Overall, the amounts of immunoblotted proteins were under the levels of detection (or lost) in the cell lysates from the group that was treated with 100 mg/dL cholesterol, therefore this group was not included in densitometric quantification (Figure 3(A)). Comparing to the control group (0 mg/dL), levels of holo-APP proteins were increased in the 12.5 and 25 mg/dL groups but reduced in the 50 mg/dL group (P < 0.0001, F = 66.0, df = 3, 8), with post-hoc tests indicating difference for the 12.5 vs. the 25 and the 50 mg/dL groups, and for the 25 vs. 50 mg/dL groups (Figure 3(B)). BACE1 protein levels were elevated in the 12.5 and 50 mg/dL groups relative to G. Z. Huang et al. 27 Figure 1. Expression of amyloidogenic proteins and Aβ production in cultured human umbilical vein endothelium cells (HUVECs). Panels (A)-(I) shows HUVECs grown in vitro 8 days after an initial expansion from the cell line, appearing in fusiform and polygonal profiles, and they express the endothelial signature protein CD31 (A) (D) (G). These cells also exhibit immunofluorescence for β-amyloid precursor protein (APP) (with the C-terminal antibody CT20), β-secretase 1 (BACE1) and presenilin-1 (PS1) (with the PS1-NTF antibody ab14). In cell lysates (J), the holo-APP proteins in HUVECs migrate at a slightly heavier molecular position relative to the counterpart immunoblotted in human brain cortical lysates. BACE1 and APP β-cleavage C-terminal fragment (APP β-CTF) are present in both the brain and HUVECs lysates (J). Note that the amount of β-actin is comparable between the two types of lysate, while the quantity of β-tubulin is minimal in the HUVECs lysates relative to human cortical samples used as assay control (J). The amount of Aβ40 in the media assayed by ELISA increases with extension of the culture days (d) (K). Insert in (K) shows a linear relationship between Aβ concentrations and light absorbance in ELISA. Scale bar = 100 μm in (A), applying to (B)-(I). G. Z. Huang et al. 28 Figure 2. Morphological characterization of the effect of cholesterol exposure on viability of cultured human umbilical vein endothelium cells (HUVECs). HUVECs are cultured using the same batch of cells for 4 days, followed by 48 hour treatment of cholesterol at 0 ((A), (A’)), 12.5 ((B), (B’)), 25 ((C), (C’)), 50 ((D), (D’)) and 100 ((E), (E)’) mg/dL concentrations and examined at the end of treatments under phase contrast microscope. The cells in the 0 - 25 mg/dL groups are healthy-looking and grown over the coverslippers ((A)-(C), (A’)-(C’)). Cells appear greatly reduced or essentially lost in the groups with 50 and 100 mg/dL cholesterol treatment. ((D), (E), (D’), (E’)). Panel (F) shows the relative viability scores with data normalized to the 0 mg/dL control group (defined as 100%), based on 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) fluogenic assay. There is a dose-dependent decline in cell viability (P < 0.0001, F = 32.6, df = 4, 10, one-way ANOVA), with posthoc test showing significant difference (* P < 0.05) in the 25 to 100 mg/dL groups relative to control. control (P < 0.0001, F = 17.3, df = 3, 8), showing difference for the 0 vs. the 25 mg/dL and for the 12.5 and 25 vs. the 50 mg/dL groups by post-hoc test (Figure 3(C)). Levels of APP β-CTF tended to increase in the 12.5 and 24 mg/dL groups but reduce in the 50 mg/dL group relative to control (P < 0.0001, F = 57.1, df = 3, 8), with significant difference between each pair of comparing dosage groups except for the 0 vs. 12.5 mg/dL groups (Figure 3(D)). Furthermore, APP β-site cleavage enzymatic activity exhibited a trend of increase in the 12.5 and 50 mg/dL groups and a reduction in the 100 mg/dL group relative to control (P < 0.0001, F = 32.8, df = 4, 10). Post-hoc test reported significant difference for the 0 vs. 25 mg/dL, the 12.5 vs. the 25 and 100 mg/dL groups, the 25 vs. 50 and 100 mg/dL and the 50 vs. 100 mg/dL groups (Figure 3(E)). Finally, Aβ40 levels in the media were elevated among the cholesterol treatment groups relative to control except for the 100 mg/dL group (P < 0.0001, F = 31.0, df = 4, 10). There existed significant difference for the control relative to the 25 and 50 mg/dL groups, for the 12.5 relative to the 25 and the 50 mg/dL groups, for the 25 relative to the 50 and 100 mg/dL groups by post-hoc test (Figure 3(F)). 4. Discussion 4.1. Vascular Endothelial Aβ Genesis and Its Potential Pharmacological Implications Earlier studies have demonstrated that several vascular endothelial cell lines, including the HUVECs and brain microvascular endothelial cells (BMECs), express the substrate and enzymes for Aβ production [35]-[39]. These findings are supported by in vivo observations from mouse and human brain preparations lately [40]-[42]. We have shown that APP, β-CTF (C99), BACE1 and PS1 protein products are detectable in primary culture of isolated human arterial endothelial cells [41]. According to an early report, APP770 is the major variant of the APP isoforms expressed at the protein levels by vascular endothelial cells, whereas neurons largely express APP695 G. Z. Huang et al. 29 Figure 3. Biochemical characterization of the effect of cholesterol exposure on amyloidogenic capability of cultured human umbilical vein endothelium cells (HUVECs). Panel (A) shows immunoblot images of cell lysate proteins from one set of experiments, documenting a loss of proteins in the 100 mg/dL group, with the amounts of APP, BACE1 and APP β-CTF, but not PS1-NTF, noticeably increased in the 12.5 and 25 mg/dL groups relative to control. Quantitatively, levels APP (B), BACE1 (C) and APP β-CTF (D) are significantly elevated (* ) or tended to increase (# ) in these two groups as compared to control. β-site APP cleavage (BACE1) enzymatic activity of the cell lysate is higher in the 12.5 - 50 mg/dL groups relative to control, with the 25 mg/dL group exhibiting statistical significance (E). Aβ40 levels in the media are elevated in the 12.5 - 50 mg/dL groups relative to control (F). * P < 0.05 relative to control. [37]. Consistent with this notion, the holo-APP proteins immunoblotted in primary human endothelial cultures migrate at a slightly heavier molecular weight position relative to its counterpart from brain lysates [41]. Importantly, data from humans and transgenic mouse models of AD suggest that Aβ overproduction in cerebral vascular endothelia, potentially mediated by excessive BACE1 activity, may contribute to the development of CAA [40]-[42], especially at the early pathogenic stage [41]. Here we have verified morphologically and biochemically that cultured HUVECs possess the full set of biochemical machinery of amyloidogenesis. Thus, these cells exhibit immunolabeling for APP and BACE1, as well as PS1 that serves the catalytic subunit of the γ-secretase complex [22] [55]. As with primary human vascular endothelial cultures [41], the holo-APP proteins immunoblotted with HUVECs lysates are slightly heavier than brain-derived APP, suggesting that the former likely represents the APP770 variant [37]. Moreover, Cultured HUVECs can secrete Aβ peptides, which accumulate in the media with time, as indicated by the increase of G. Z. Huang et al. 30 Aβ40 levels assessed by ELISA over different periods of cultivation. The finding that vascular endothelial cells produce Aβ may have some implications especially with regard to the design of drugs to antagonize brain amyloid pathogenesis. Attenuating Aβ overproduction by BACE1 and γ-secretase inhibitors/modulators has been explored as an pharmacological option for AD or cerebral amyloid pathology, although clinical trials on a number of γ-secretase inhibitors have failed to establish therapeutic benefits so far [56]. BACE1 inhibitors appear to be promising given that they block the initial step of the amyloidogenic pathway, and can reduce the levels of Aβ as well as APP β-CTF, the latter is also considered to be neurotoxic [18] [21] [57]-[60]. However, emerging evidence also raises concern as to whether inhibition of neuronal BACE1 may cause serious side-effects by disrupting the neurobiological role of the enzyme [61]. It is of interest to note that initially developed BACE1 inhibitors are generally not advanced into clinical trials because they are incapable of passing across the Blood Brain Barrier (BBB). In this regard, targeting endothelial Aβ genesis with brain impermeable BACE1 inhibitors may be not only pharmacologically feasible but turn out to be a blessing strategy [42]. Hypothetically, such inhibitors could act against the early rise of Aβ in vascular wall, protect BBB integrity and have less neuronal toxic effect, and therefore, may serve as preventive agents. 4.2. Cholesterol as an Inductive Factor for Vascular Aβ Overproduction Cholesterol has been long considered a risk factor for vascular dementia and AD according to epidemiologic data [43]-[53]. Cholesterol is transported between tissues/cells via apolipoprotein E and its interaction with its cell-surface receptors [46]. Inpiduals carrying the apolipoprotein E epsilon 4 allele (APOE-ε4) are shown to have an increased vulnerability to AD and dementia [48] [51]. These carries are associated with hypercholesterolemia [43] [48] [53] and increased prevalence and severity of cerebral amyloid pathology, especially CAA [2] [11] [52] [62]. Statins, which lower blood cholesterol levels, appear to be beneficial for preventing AD and dementia [49] [63]. Given vascular endothelia being positioned at the first front to potential circulatory stress factors; it is of interest to identify such factors that might affect the amyloidogenic activity in these cells. As elaborated above, cholesterol represents one of the candidate circulatory stress factors of interest to explore its influence on endothelial Aβ genesis. In the present study we find that cultured HUVECs respond to cholesterol exposure in a dose-dependent biphasic mode. At the lower doses tested in vitro, from approximately 12.5 to 25 mg/dL, cholesterol exposure potentiates amyloidogenesis in HUVECs by up-regulating APP and BACE1, but not PS1, expression. At higher concentrations, especially at 50 and 100 mg/dL, cholesterol exposure reduces the viability and causes death of the HUVECs. The cellular toxicity induced by high cholesterol exposure is associated with diminished expression of the amyloidogenic (reduce or loss of APP, BACE1 and β-CTF immunoblotting signal) and house-keeping proteins (loss of GADPH immunoblotting signal), and reduced Aβ production (lowering of Aβ levels in culture medium), in the HUVECs. These findings imply that elevated blood cholesterol could be a factor to potentiate amyloidogenesis in vascular endothelial cells. In clinical practise, hypercholesterolemia is often defined by blood total cholesterol concentration greater than 200 mg/dL (this cutoff may vary from hospital to hospital). In the current in vitro setting, cultured HUVECs appeared to be very sensitive to exogenous cholesterol challenge. Indeed, at concentrations much lower than 200 mg/dL, they already responded to cholesterol exposure quite robustly (from enhanced Aβ genesis and even to compromised whole cell viability). The dose-effect response observed in our in vitro experiments was progressive relative to the cholesterol concentrations tested. Notably, the low concentration treatments (12.5 mg/dL as well as 6.25 mg/dL tested in pilot studies) did not apparently impair cell morphology, viability or responses of protein (e.g., APP, BACE1 or β-actin) expression. Therefore, while our findings could (and should) not be truly correlated to hypercholesterolemia in a clinical term (perhaps as it is always for the case of in vitro experiments), the data clearly demonstrate a modulatory effect of cholesterol on amyloidogenesis in vascular endothelial cells. Translation of in vitro findings towards clinical relevance needs to be always cautious. Bearing an interpretative limitation in mind, the results obtained in this study are nonetheless relevant to the understanding of a potential pathophysiological role of hypercholesterolemia. 5. Conclusion In summary, cultured human umbilical vein endothelial cells express a full set of biochemical machinery for the production of β-amyloid peptides and are capable of secreting the peptides into culture medium. The amyloido- G. Z. Huang et al. 31 genic activity of these cells is enhanced in response to the exposure of cholesterol within a certain concentration range, while strong cholesterol challenge deteriorates their viability. These findings point to a possibility that hypercholesterolemia may serve as a circulatory factor to promote β-amyloid production in and release from vascular endothelial cells. Acknowledgements We thank Edward Koo, Huaibin Cai and Samuel E Gandy for providing the APP, BACE1 and PS1-NTF antibodies. This study was supported by National Natural Science Foundation of China (#81171160, #31371095) and approved by the Ethical Committee for biomedical research at Xiangya School of Medicine. The authors declare that the research was conducted in the absence of any commercial or financial relationships. References [1] Castellani, R.J., Smith, M.A., Perry, G. and Friedland, R.P. (2004) Cerebral Amyloid Angiopathy: Major Contributor or Decorative Response to Alzheimer’s Disease Pathogenesis. Neurobiology Aging, 25, 599-602. http://dx.doi.org/10.1016/j.neurobiolaging.2003.12.019 [2] Thal, D.R., Griffin, W.S., de Vos, R.A. and Ghebremedhin, E. (2008) Cerebral Amyloid Angiopathy and Its Relationship to Alzheimer’s Disease. Acta Neuropathologica, 115, 599-609. http://dx.doi.org/10.1007/s00401-008-0366-2 [3] Weller, R.O., Boche, D. and Nicoll, J.A. (2009) Microvasculature Changes and Cerebral Amyloid Angiopathy in Alzheimer’s Disease and Their Potential Impact on Therapy. Acta Neuropathologica, 118, 87-102. http://dx.doi.org/10.1007/s00401-009-0498-z [4] Honjo, K., Black, S.E. and Verhoeff, N.P. (2012) Alzheimer’s Disease, Cerebrovascular Disease, and the β-Amyloid Cascade. Canadian Journal of Neurological Sciences, 39, 712-728. http://dx.doi.org/10.1017/S0317167100015547 [5] Pfeifer, L.A., White, L.R., Ross, G.W., Petrovitch, H. and Launer, L.J. (2002) Cerebral Amyloid Angiopathy and Cognitive Function: The HAAS Autopsy Study. Neurology, 58, 1629-1634. http://dx.doi.org/10.1212/WNL.58.11.1629 [6] Jellinger, K.A. and Attems, J. (2003) Incidence of Cerebrovascular Lesions in Alzheimer’s Disease: A Postmortem Study. Acta Neuropathologica, 105, 14-17. [7] Keage, H.A., Carare, R.O., Friedland, R.P., Ince, P.G., Love, S., Nicoll, J.A., Wharton, S.B., Weller, R.O. and Brayne, C. (2009) Population Studies of Sporadic Cerebral Amyloid Angiopathy and Dementia: A Systematic Review. BMC Neurology, 9, 3. http://dx.doi.org/10.1186/1471-2377-9-3 [8] Arvanitakis, Z., Leurgans, S.E., Wang, Z., Wilson, R.S., Bennett, D.A. and Schneider, J.A. (2011) Cerebral Amyloid Angiopathy Pathology and Cognitive Domains in Older Persons. Annals of Neurology, 69, 320-327. http://dx.doi.org/10.1002/ana.22112 [9] Obrien, R.J. (2011) Vascular Dementia: Atherosclerosis, Cognition and Alzheimer’s Disease. Current Alzheimer Research, 8, 341-344. http://dx.doi.org/10.2174/156720511795745267 [10] Viswanathan, A. and Greenberg, S.M. (2011) Cerebral Amyloid Angiopathy in the Elderly. Annals of Neurology, 70, 871-880. http://dx.doi.org/10.1002/ana.22516 [11] Purrucker, J.C., Hund, E., Ringleb, P.A., Hartmann, C., Rohde, S., Schönland, S. and Steiner, T. (2013) Cerebral Amyloid Angiopathy—An Underdiagnosed Entity in Younger Adults with Lobar Intracerebral Hemorrhage? Amyloid, 20, 45-47. http://dx.doi.org/10.3109/13506129.2012.746937 [12] Charidimou, A., Gang, Q. and Werring, D.J. (2012) Sporadic Cerebral Amyloid Angiopathy Revisited: Recent Insights into Pathophysiology and Clinical Spectrum. Journal of Neurology, Neurosurgery, and Psychiatry, 83, 124-137. http://dx.doi.org/10.1136/jnnp-2011-301308 [13] Allen, N., Robinson, A.C., Snowden, J., Davidson, Y.S. and Mann, D.M. (2014) Patterns of Cerebral Amyloid Angiopathy Define Histopathological Phenotypes in Alzheimer’s Disease. Neuropathology and Applied Neurobiology, 40, 136-148. http://dx.doi.org/10.1111/nan.12070 [14] Geddes, J.W. and Cotman, C.W. (1991) Plasticity in Alzheimer’s Disease: Too Much or Not Enough? Neurobiology of Aging, 12, 330-333. http://dx.doi.org/10.1016/0197-4580(91)90011-8 [15] Arendt, T. (2001) Alzheimer’s Disease as a Disorder of Mechanisms Underlying Structural Brain Self-Organization. Neuroscience, 102, 723-765. http://dx.doi.org/10.1016/S0306-4522(00)00516-9 [16] Castellani, R.J., Lee, H.G., Zhu, X., Perry, G. and Smith, M.A. (2008) Alzheimer Disease Pathology as a Host Response. Journal of Neuropathology and Experimental Neurology, 67, 523-531. http://dx.doi.org/10.1097/NEN.0b013e318177eaf4 G. Z. Huang et al. 32 [17] Li, J.M., Liu, C., Hu, X., Cai, Y., Ma, C., Luo, X.G. and Yan, X.X. (2014) Inverse Correlation between Alzheimer’s Disease and Cancer: Implication for a Strong Impact of Regenerative Propensity on Neurodegeneration? BMC Neurology, 14, 211. http://dx.doi.org/10.1186/s12883-014-0211-2 [18] Yan, X.X., Ma, C., Gai, W.P., Cai, H. and Luo, X.G. (2014) Can BACE1 Inhibition Mitigate Early Axonal Pathology in Neurological Diseases? Journal of Alzheimer’s Disease, 38, 705-718 [19] Li, J.M., Cai, Y., Liu, F., Yang, L., Hu, X., Patrylo, P.R., Cai, H., Luo, X.G., Xiao, D. and Yan, X.X. (2015) Experimental Microembolism Induces Localized Neuritic Pathology in Guinea Pig Cerebrum. Oncotarget, 6, 10772-10785. http://dx.doi.org/10.18632/oncotarget.3599 [20] Laird, F.M., Cai, H., Savonenko, A.V., Farah, M.H., He, K., Melnikova, T., Wen, H., Chiang, H.C., Xu, G., Koliatsos, V.E., Borchelt, D.R., Price, D.L., Lee, H.K. and Wong, P.C. (2005) BACE1, a Major Determinant of Selective Vulnerability of the Brain to Amyloid-Beta Amyloidogenesis, Is Essential for Cognitive, Emotional, and Synaptic Functions. Journal of Neuroscience, 25, 11693-11709. http://dx.doi.org/10.1523/JNEUROSCI.2766-05.2005 [21] Vassar, R., Kovacs, D.M., Yan, R. and Wong, P.C. (2009) The Beta-Secretase Enzyme BACE in Health and Alzheimer’s Disease: Regulation, Cell Biology, Function, and Therapeutic Potential. Journal of Neuroscience, 29, 12787- 12794. http://dx.doi.org/10.1523/JNEUROSCI.3657-09.2009 [22] Liu, F., Xue, Z.Q., Deng, S.H., Kun, X., Luo, X.G., Patrylo, P.R., Rose, G.M., Cai, H., Struble, R.G., Cai, Y. and Yan, X.X. (2013) γ-Secretase Binding Sites in Aged and Alzheimer’s Disease Human Cerebrum: the Choroid Plexus as a Putative Origin of CSF Aβ. European Journal of Neuroscience, 37, 1714-1725. http://dx.doi.org/10.1111/ejn.12159 [23] Holsinger, R.M., McLean, C.A., Beyreuther, K., Masters, C.L. and Evin, G. (2002) Increased Expression of the Amyloid Precursor Beta-Secretase in Alzheimer’s Disease. Annals of Neurology, 51, 783-786. http://dx.doi.org/10.1002/ana.10208 [24] Tyler, S.J., Dawbarn, D., Wilcock, G.K. and Allen, S.J. (2002) Alpha- and Beta-Secretase: Profound Changes in Alzheimer’s Disease. Biochemical and Biophysical Research Communications, 299, 373-376. http://dx.doi.org/10.1016/S0006-291X(02)02635-9 [25] Fukumoto, H., Cheung, B.S., Hyman, B.T. and Irizarry, M.C. (2002) Beta-Secretase Protein and Activity are Increased in the Neocortex in Alzheimer Disease. Archives of Neurology, 59, 1381-1389. http://dx.doi.org/10.1001/archneur.59.9.1381 [26] Yang, L.B., Lindholm, K., Yan, R., Citron, M., Xia, W., Yang, X.L., Beach, T., Sue, L., Wong, P., Price, D., Li, R. and Shen, Y. (2003) Elevated Beta-Secretase Expression and Enzymatic Activity Detected in Sporadic Alzheimer Disease. Nature Medicine, 9, 3-4. http://dx.doi.org/10.1038/nm0103-3 [27] Li, R., Lindholm, K., Yang, L.B., Yue, X., Citron, M., Yan, R., Beach, T., Sue, L., Sabbagh, M., Cai, H., Wong, P., Price, D. and Shen, Y. (2004) Amyloid Beta Peptide Load Is Correlated with Increased Beta-Secretase Activity in Sporadic Alzheimer’s Disease Patients. Proceedings of the National Academy of Sciences of the United States of America, 101, 3632-3637. http://dx.doi.org/10.1073/pnas.0205689101 [28] Coulson, D.T., Beyer, N., Quinn, J.G., Brockbank, S., Hellemans, J., Irvine, G.B., Ravid, R. and Johnston, J.A. (2010) BACE1 mRNA Expression in Alzheimer’s Disease Postmortem Brain Tissue. Journal of Alzheimer’s Disease, 22, 1111-1122. [29] Cai, Y., Xiong, K., Zhang, X.M., Cai, H., Luo, X.G., Feng, J.C., Clough, R.W., Struble, R.G., Patrylo, P.R., Chu, Y., Kordower, J.H. and Yan, X.X. (2010) β-Secretase-1 Elevation in Aged Monkey and Alzheimer’s Disease Human Cerebral Cortex Occurs around the Vasculature in Partnership with Multisystem Axon Terminal Pathogenesis and β-Amyloid Accumulation. European Journal of Neuroscience, 32, 1223-1238. http://dx.doi.org/10.1111/j.1460-9568.2010.07376.x [30] Cheng, X., He, P., Lee, T., Yao, H., Li, R. and Shen, Y. (2014) High Activities of BACE1 in Brains with Mild Cognitive Impairment. American Journal of Pathology, 184, 141-147. http://dx.doi.org/10.1016/j.ajpath.2013.10.002 [31] Zhao, J., Fu, Y., Yasvoina, M., Shao, P., Hitt, B., O’Connor, T., Logan, S., Maus, E., Citron, M., Berry, R., Binder, L. and Vassar, R. (2007) Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 Levels Become Elevated in Neurons around Amyloid Plaques: Implications for Alzheimer’s Disease Pathogenesis. Journal of Neuroscience, 27, 3639-3649. http://dx.doi.org/10.1523/JNEUROSCI.4396-06.2007 [32] Zhang, X.M., Cai, Y., Cai, H., Xiong, K., Luo, X.G., Feng, J.C., Clough, R.W., Struble, R.G., Patrylo, P.R. and Yan, X.X. (2009) BACE1 Elevation in Transgenic Mouse Models of Alzheimer’s Disease Is Associated with Synaptic/Axonal Pathology and Amyloidogenesis: Implications for Neuritic Plaque Development. European Journal of Neuroscience, 30, 2271-2283. http://dx.doi.org/10.1111/j.1460-9568.2009.07017.x [33] Cai, Y., Zhang, X.M., Macklin, L.N., Cai, H., Luo, X.G., Oddo, S., Laferla, F.M., Struble, R.G., Rose, G.M., Patrylo, P.R. and Yan, X.X. (2012) BACE1 Elevation Is Involved in Amyloid Plaque Development in the Triple Transgenic Model of Alzheimer’s Disease: Differential Aβ Antibody Labeling of Early-Onset Axon Terminal Pathology. Neurotoxicity Research, 21, 160-174. http://dx.doi.org/10.1007/s12640-011-9256-9 G. Z. Huang et al. 33 [34] Kandalepas, P.C., Sadleir, K.R., Eimer, W.A., Zhao, J., Nicholson, D.A. and Vassar, R. (2013) The Alzheimer’s β-Secretase BACE1 Localizes to Normal Presynaptic Terminals and to Dystrophic Presynaptic Terminals Surrounding Amyloid Plaques. Acta Neuropathologica, 126, 329-352. http://dx.doi.org/10.1007/s00401-013-1152-3 [35] Simons, E.R., Marshall, D.C., Long, H.J., Otto, K., Billingslea, A., Tibbles, H., Wells, J., Eisenhauer, P., Fine, R.E., Cribbs, D.H., Davies, T.A. and Abraham, C.R. (1998) Blood Brain Barrier Endothelial Cells Express Candidate Amyloid Precursor Protein-Cleaving Secretases. Amyloid, 5, 153-162. http://dx.doi.org/10.3109/13506129809003841 [36] Austin, S.A., Santhanam, A.V. and Katusic, Z.S. (2010) Endothelial Nitric Oxide Modulates Expression and Processing of Amyloid Precursor Protein. Circulation Research, 107, 1498-502. http://dx.doi.org/10.1161/CIRCRESAHA.110.233080 [37] Kitazume, S., Tachida, Y., Kato, M., Yamaguchi, Y., Honda, T., Hashimoto, Y., Wada, Y., Saito, T., Iwata, N., Saido, T. and Taniguchi, N. (2010) Brain Endothelial Cells Produce Amyloid β from Amyloid Precursor Protein 770 and Preferentially Secrete the O-Glycosylated Form. The Journal of Biological Chemistry, 285, 40097-40103. http://dx.doi.org/10.1074/jbc.M110.144626 [38] Ma, J.F., Wang, H.M., Li, Q.Y., Zhang, Y., Pan, J., Qiang, Q., Xin, X.Y., Tang, H.D., Ding, J.Q. and Chen, S.D. (2010) Starvation Triggers Abeta42 Generation from Human Umbilical Vascular Endothelial Cells. FEBS Letters, 584, 3101- 3106. http://dx.doi.org/10.1016/j.febslet.2010.05.048 [39] Bulbarelli, A., Lonati, E., Brambilla, A., Orlando, A., Cazzaniga, E., Piazza, F., Ferrarese, C., Masserini, M. and Sancini, G. (2012) Aβ42 Production in Brain Capillary Endothelial Cells after Oxygen and Glucose Deprivation. Molecular and Cellular Neuroscience, 49, 415-422. http://dx.doi.org/10.1016/j.mcn.2012.01.007 [40] Cheng, X., He, P., Yao, H., Dong, Q., Li, R. and Shen, Y. (2014) Occludin Deficiency with BACE1 Elevation in Cerebral Amyloid Angiopathy. Neurology, 82, 1707-1715. http://dx.doi.org/10.1212/WNL.0000000000000403 [41] Xue, Z.Q., He, Z.W., Yu, J.J., Cai, Y., Qiu, W.Y., Pan, A., Gai, W.P., Cai, H., Luo, X.G., Ma, C. and Yan, X.X. (2015) Non-Neuronal and Neuronal BACE1 Elevation in Association with Angiopathic and Leptomeningeal β-Amyloid Deposition in the Human Brain. BMC Neurology, 15, 71. http://dx.doi.org/10.1186/s12883-015-0327-z [42] Devraj, K., Poznanovic, S., Spahn, C., Schwall, G., Harter, P.N., Mittelbronn, M., Antoniello, K., Paganetti. P., Muhs, A., Heilemann, M., Hawkins, R.A., Schrattenholz, A. and Liebner, S. (2015) BACE-1 Is Expressed in the Blood-Brain Barrier Endothelium and Is Upregulated in a Murine Model of Alzheimer’s Disease. Journal of Cerebral Blood Flow and Metabolism. http://dx.doi.org/10.1177/0271678x15606463 [43] Utermann, G., Kindermann, I., Kaffarnik, H. and Steinmetz, A. (1984) Apolipoprotein E Phenotypes and Hyperlipidemia. Human Genetics, 65, 232-236. http://dx.doi.org/10.1007/BF00286508 [44] Li, G., Shofer, J.B., Kukull, W.A., Peskind, E.R., Tsuang, D.W., Breitner, J.C., McCormick, W., Bowen, J.D., Teri, L., Schellenberg, G.D. and Larson, E.B. (2005) Serum Cholesterol and Risk of Alzheimer Disease: A Community-Based Cohort Study. Neurology, 65, 1045-1050. http://dx.doi.org/10.1212/01.wnl.0000178989.87072.11 [45] Anstey, K.J., Lipnicki, D.M. and Low, L.F. (2008) Cholesterol as a Risk Factor for Dementia and Cognitive Decline: A Systematic Review of Prospective Studies with Meta-Analysis. American Journal of Geriatric Psychiatry, 16, 343- 354. http://dx.doi.org/10.1097/01.JGP.0000310778.20870.ae [46] Mahley, R.W., Huang, Y. and Weisgraber, K.H. (2006) Putting Cholesterol in Its Place: ApoE and Reverse Cholesterol Transport. Journal of Clinical Investigation, 116, 1226-1229. http://dx.doi.org/10.1172/JCI28632 [47] Solomon, A., Kivipelto, M., Wolozin, B., Zhou, J. and Whitmer, R.A. (2009) Midlife Serum Cholesterol and Increased Risk of Alzheimer’s and Vascular Dementia Three Decades Later. Dementia and Geriatric Cognitive Disorder, 28, 75- 80. http://dx.doi.org/10.1159/000231980 [48] Leduc, V., Jasmin-Bélanger, S. and Poirier, J. (2010) APOE and Cholesterol Homeostasis in Alzheimer’s Disease. Trends in Molecular Medicine, 16, 469-477. http://dx.doi.org/10.1016/j.molmed.2010.07.008 [49] Shepardson, N.E., Shankar, G.M. and Selkoe, D.J. (2011) Cholesterol Level and Statin Use in Alzheimer Disease: II. Review of Human Trials and Recommendations. Archives of Neurology, 68, 1385-1392. http://dx.doi.org/10.1001/archneurol.2011.242 [50] Tolppanen, A.M., Solomon, A., Soininen, H. and Kivipelto, M. (2012) Midlife Vascular Risk Factors and Alzheimer’s Disease: Evidence from Epidemiological Studies. Journal of Alzheimer’s Disease, 32, 531-540. [51] Sadigh-Eteghad, S., Talebi, M. and Farhoudi, M. (2012) Association of Apolipoprotein E Epsilon 4 Allele with Sporadic Late Onset Alzheimer’s Disease. A Meta-Analysis. Neurosciences (Riyadh), 17, 321-326. [52] Silva, T., Teixeira, J., Remião, F. and Borges, F. (2013) Alzheimer’s Disease, Cholesterol, and Statins: The Junctions of Important Metabolic Pathways. Angewandte Chemie International Edition in English, 52, 1110-1121. http://dx.doi.org/10.1002/anie.201204964 [53] Reed, B., Villeneuve, S., Mack, W., Decarli, C., Chui, H.C. and Jagust, W. (2014) Associations between Serum Cholesterol Levels and Cerebral Amyloidosis. JAMA Neurology, 71, 195-200. G. Z. Huang et al. 34 http://dx.doi.org/10.1001/jamaneurol.2013.5390 [54] Maulik, M., Westaway, D., Jhamandas, J.H. and Kar, S. (2013) Role of Cholesterol in APP Metabolism and Its Significance in Alzheimer’s Disease Pathogenesis. Molecular Neurobiology, 47, 37-63. http://dx.doi.org/10.1007/s12035-012-8337-y [55] Yan, X.X., Li, T., Rominger, C.M., Prakash, S.R., Wong, P.C., Olson, R.E., Zaczek, R. and Li, Y.W. (2004) Binding Sites of Gamma-Secretase Inhibitors in Rodent Brain: Distribution, Postnatal Development, and Effect of Deafferentation. The Journal of Neuroscience, 24, 2942-2952. http://dx.doi.org/10.1523/JNEUROSCI.0092-04.2004 [56] De Strooper, B. and Chávez Gutiérrez, L. (2015) Learning by Failing: Ideas and Concepts to Tackle γ-Secretases in Alzheimer’s Disease and beyond. Annual Review of Pharmacology and Toxicology, 55, 419-437. http://dx.doi.org/10.1146/annurev-pharmtox-010814-124309 [57] Kim, H.S., Park, C.H., Cha, S.H., Lee, J.H., Lee, S., Kim, Y., Rah, J.C., Jeong, S.J. and Suh, Y.H. (2000) Carboxyl-Terminal Fragment of Alzheimer’s APP Destabilizes Calcium Homeostasis and Renders Neuronal Cells Vulnerable to Excitotoxicity. The FASEB Journal, 14, 1508-1517. http://dx.doi.org/10.1096/fj.14.11.1508 [58] Saganich, M.J., Schroeder, B.E., Galvan, V., Bredesen, D.E., Koo, E.H. and Heinemann, S.F. (2006) Deficits in Synaptic Transmission and Learning in Amyloid Precursor Protein (APP) Transgenic Mice Require C-Terminal Cleavage of APP. The Journal of Neuroscience, 26, 13428-13436. http://dx.doi.org/10.1523/JNEUROSCI.4180-06.2006 [59] Lauritzen, I., Pardossi-Piquard, R., Bauer, C., Brigham, E., Abraham, J.D., Ranaldi, S., Fraser, P., St-George-Hyslop, P., Le Thuc, O., Espin, V., Chami, L., Dunys, J. and Checler, F. (2012) The β-Secretase-Derived C-Terminal Fragment of βAPP, C99, but Not Aβ, Is a Key Contributor to Early Intraneuronal Lesions in Triple-Transgenic Mouse Hippocampus. The Journal of Neuroscience, 32, 16243-16255. http://dx.doi.org/10.1523/JNEUROSCI.2775-12.2012 [60] Jiang, Y., Rigoglioso, A., Peterhoff, C.M., Pawlik, M., Sato, Y., Bleiwas, C., Stavrides, P., Smiley, J.F., Ginsberg, S.D., Mathews, P.M., Levy, E. and Nixon, R.A. (2016) Partial BACE1 Reduction in a Down Syndrome Mouse Model Blocks Alzheimer-Related Endosomal Anomalies and Cholinergic Neurodegeneration: Role of APP-CTF. Neurobiology of Aging, 39, 90-98. http://dx.doi.org/10.1016/j.neurobiolaging.2015.11.013 [61] Barão, S., Moechars, D., Lichtenthaler, S.F. and De Strooper, B. (2016) BACE1 Physiological Functions May Limit Its Use as Therapeutic Target for Alzheimer’s Disease. Trends in Neurosciences, 39, 158-269. http://dx.doi.org/10.1016/j.tins.2016.01.003 [62] Nicoll, J.A. and McCarron, M.O. (2001) APOE Gene Polymorphism as a Risk Factor for Cerebral Amyloid Angiopathy-Related Hemorrhage. Amyloid, 8, 51-55. [63] Kurata, T., Kawai, H., Miyazaki, K., Kozuki, M., Morimoto, N., Ohta, Y., Ikeda, Y. and Abe, K. (2012) Statins Have Therapeutic Potential for the Treatment of Alzheimer’s Disease, Likely via Protection of the Neurovascular Unit in the AD Brain. Journal of the Neurological Sciences, 322, 59-63. http://dx.doi.org/10.1016/j.jns.2012.06.011
Cholesterol Potentiates β-Amyloid Genesis in Cultured Human Umbilical Vein Endothelial Cells Guozhi Huang1,2, Zhiqin Xue3, Xia Hu1, Lily Wan1, Jianming Li4, Yan Cai1, Peter R. Patrylo5, Xuegang Luo1, Aihua Pan1, Xiao-Xin Yan1,6* 1 Department of Anatomy and Neurobiology, Central South University Xiangya Medical School, Changsha, China 2 Department of Anatomy & Embryology, Loudi Vocational and Technical College, Loudi, China 3 Department of Anatomy, Xinjiang Medical University, Urumqi, China 4 Neuroscience Research Center, Changsha Medical University, Changsha, China 5 Departments of Physiology, Anatomy and the Center for Integrated Research in Cognitive and Neural Sciences, Southern Illinois University Carbondale, Carbondale, USA 6 Key Laboratory of Hunan Province in Neurodegenerative Disorders, Changsha, China Received 20 April 2016; accepted 3 June 2016; published 6 June 2016 Copyright © 2016 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Cerebral Amyloid Angiopathy (CAA) occurs commonly among the elderly and almost invariably in patients with Alzheimer’s Disease (AD). The β-amyloid peptides (Aβ) are produced via the amyloidogenic processing of β-Amyloid Precursor Protein (APP) by β-secretase-1 (BACE1) and γ- secretase. Vascular endothelial cells are lately shown to possess the molecular machinery of Aβ production, which might participate in the development of CAA. Hypercholesterolemia is considered a risk factor for AD, whereas less is known if cholesterol may modulate endothelial Aβ production. In the present study we verified the amyloidogenic capability of Human Umbilical Vein Endothelial Cells (HUVECs) in vitro and explored the effect of cholesterol exposure on their amyloidogenic potential. Cholesterol treatments at 12.5 and 25 mg/dL significantly elevated APP, BACE1 and APP β-CTF protein levels and β-site APP cleavage activity in cell lysates, and Aβ40 levels in culture medium. However, coincubation with cholesterol at 50 and 100 mg/dL attenuated the viability of the cultured cells and diminished their amyloidogenic capability. These findings suggest that high cholesterol exposure is stressful to vascular endothelial cells, and at a certain dosage range can promote an amyloidogenic response in these cells. * Corresponding author. G. Z. Huang et al. 24 Keywords Aging, Alzheimer’s Disease, Cholesterol, Neurodegeneration, Vascular Dementia 1. Introduction Cerebral amyloidosis is a pathological condition caused by the deposition of the β-amyloid peptides (Aβ) in the extracellular matrix and vascular wall in the central nervous system, referred to as parenchymal amyloid plaques and Cerebral Amyloid Angiopathy (CAA), respectively [1] [2]. Amyloid plaques and CAA are among the definitive neuropathological lesions of dementia of the Alzheimer’s Disease (AD) and vascular types [3]-[5], although both types of amyloidosis can be seen in the brain of non-demented elderly [6]-[13]. Along with Aβ deposits, parenchymal and vascular amyloid pathology may coexist with degenerative and reactive cellular changes, including neuronal and vascular cell loss, neuritic dystrophy and glial activation [14]-[19]. Neurons appear to be the major origin of soluble and insoluble Aβ products in the brain given that they are enriched with the amyloidogenic biochemical machinery including β-Amyloid Precursor Protein (APP), β-secretatse-1 (BACE1) and γ-secretase [20]-[22]. In particular, elevated BACE1 protein and enzymatic activity are reported in the human brains with amyloid neuropathology [22]-[30]. Specifically, enhanced BACE1 expression is identified microscopically in dystrophic axons around neuritic and angiopathic Aβ deposition [18] [31]-[34]. Vascular endothelial cells are initially shown to express the full set of amyloidogenic machinery in vitro, which has been lately demonstrated in vivo including in the human brain with CAA [35]-[42]. It is suggested that Aβ overproduction by vascular endothelial cells may be involved in the early stages of the development of CAA [40]-[42]. Therefore, it becomes important to identify circulatory factors that might potentiate endothelial amyloidogenesis. Hypercholesterolemia has been considered a risk factor for AD and vascular dementia for a long time [43]-[53]. However, it remains less clear if cholesterol may directly influence the amyloidogenic activity in vascular endothelial cells [54]. In the present study, we used Human Umbilical Vein Endothelial Cells (HUVECs) as a model to explore a modulatory effect of cholesterol on endothelial amyloidogenic protein expression and Aβ secretion. Cultured HUVECs exhibited a biphasic response to cholesterol treatments, with low dose exposure potentiated Aβ genesis while high dose exposure resulted in cell death and diminished Aβ production. 2. Materials and Methods 2.1. Cell Culture and Cholesterol Treatment HUVECs were obtained commercially (Catalog #C0035C, Life Technologies Corporation). Cell supplies were re-suspended with DMEM/F12 containing 10% fetal bovine serum, to a beginning density approximately 5 × 106 cells/mL. Cells were cultured at 37˚C in a humidified atmosphere containing 95% air and 5% CO2, in six-well Corning-Costar plates (Catalog #CLS3516; Life Technologies Corporation). Each well was loaded with 2 ml of medium and a glass coverslip coated with polylysine, with the medium changed twice a week. Initial experiments were carried out to verify the cultured cells and optimize the biochemical assays. Cholesterol treatments to HUVECs were initiated on day 4 of culture for each round of experiments for duration of 48 hours. A total of 3 experiments were carried out separately in different times. For each experiment, media containing 0, 12.5, 25, 50 and 100 mg/dL of cholesterol (Catalog #C8667, Sigma-Aldrich, St. Louis, MO, USA) were added in duplicate into wells loaded with the same amount of cells from the same original batch of HUVECs expansion. Media were collected following 48 hours of culture to assess Aβ40 concentrations, with the corresponding groups of cultured cells harvested for morphological study and assessment of related proteins and APP β-cleavage activity. 2.2. Immunocytochemistry and Microscopic Examination HUVECs grown on coverslippers were fixed in situ with cold 4% paraformaldehyde for 20 minutes and stored frozen until immunofluorescent processing. Coverslippers were thawed to room temperature prior to staining, and incubated in PBS containing 5% donkey serum for 30 minutes, followed by reaction with a pair of primary G. Z. Huang et al. 25 antibodies raised in different species overnight at 4˚C, including 1) mouse anti-CD31 (endothelial marker) [41] and rabbit anti-APP C-terminal (CT20); 2) mouse anti-CD31 and rabbit anti-BACE1 [20] [32]; 3) mouse antiCD31 and rabbit anti-presilinin-1 (PS1) N-terminal (ab14) [41]. Following several rinses with PBS, the coverslippers were further incubated at room temperature for a 2 hour with Alexa Fluor® 488 donkey anti-rabbit and Alexa Fluor® 594 conjugated donkey anti-mouse IgGs (1:200, Invitrogen, Carlsbad, CA, USA). Coverslippers were finally counter-stained with bisbenzimide (Hoechst 33,342, 1:50,000, Catalog #B2261, Sigma-Aldrich), washed thoroughly, and mounted with anti-fading medium. Immunofluorescence was examined on an Olympus fluorescent BX53 microscope equipped with a digital imaging system (CellSens Standard, Olympus Corporation, Japan). 2.3. Western Blot HUVECs pellets (×4, w⁄v) were homogenized by sonication in T-PER extraction buffer (Pierce, Rockford, IL, USA) containing protease inhibitors (Roche, Indianapolis, IN, USA). The lysates were centrifuged at 15,000 g, with supernatants collected and protein concentrations determined by DC protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Human cortical lysates available from a previous study (Xue et al., 2015) were used as an assay control to cross-validate the protein profile of the HUVECs relative to brain tissue. Fifty microgram protein was run on each lane in 10% or 18% (for APP β-CTF) SDS-polyacrylamide electrophoresis gels. Separated proteins were electrotransferred to Trans-Blot pure nitrocellulose membranes (Bio-Rad Lab.), which were immunoblotted with antibodies to APP (22C11), APP β-CTF (AHP538), BACE1, PS1-NTF (ab14), β-tubulin-III, β-actin at pre-optimized working dilutions with agitation for 12 hours. The membranes were further reacted with horseradish peroxidase (HRP)-conjugated goat anti-rabbit or anti-mouse IgGs (1:20,000; Bio-Rad Laboratories), treated with the ECL Plus Western Blotting Detection Kit, and exposed against Kodak X-OMAT Blue autoradiography films that were developed in a darkroom subsequently. 2.4. APP β-Site Cleavage (BACE1) Activity Assay APP β-site cleavage activity (representing BACE1 enzymatic activity) in HUVECs was measured in duplicate in 96-well transparent flat-bottomed plates. Samples were homogenized on ice as described above for Western blot, and assayed for enzymatic activity using a commercial kit (Calbiochem, La Jolla, CA, USA, Catalog #565785) according to manufacturer’s instructions. The fluorescent signal was captured in a Bio-Rad microplate reader (PR 3100 TSC). 2.5. Enzyme-Linked Immunosorbent Assay for Aβ in Culture Medium Aβ levels in cell culture media were obtained to estimate the amount of secreted peptides following cultivation for different time periods or treatments of cholesterol at different concentrations. Media were assayed in duplicate by solid sandwich enzyme-linked immunosorbent assay (ELISA) to assess Aβ40 levels using commercial kits (Novex® KHB3482 for Aβ40) following the manufacturer’s instruction (Life Technologies Corporation). Signals were captured in the Bio-Rad microplate reader, with the Aβ concentrations (pM/ml) calculated according to the standard curve generated from the readouts of the synthesized peptides supplied along with the kit. 2.6. MTT Cell Viability Assay The viability of HUVECs was assayed at the end of cholesterol treatment experiments using a commercial fluogenic 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) kit following the manufacturer’s instruction (Abnova, Catalog # KA1606, Taipei City, Taiwan). Fluorescent signal was measured in the Bio-Rad microplate reader, with cell viability calculated for comparing culture groups. 2.7. Quantification, Statistical Analysis and Figure Preparation Densitometric (Western blot, digitalized using NIH-Image J) and numeric data (light absorbent signals of APP β-site cleavage enzymatic activity, Aβ ELISA and MTT fluogenic activity) were normalized to either the internal standard or the control group. Means were calculated for comparing groups, and analyzed statistically using one-way ANOVA with Bonferroni’s multiple comparison tests (Prism GraphPad, San Diego, CA). The significant level of difference was set at P < 0.05. Figures were assembled with Photoshop 7.0, with brightness/ G. Z. Huang et al. 26 contract adjusted as needed. 3. Results 3.1. HUVECs Expressed Amyloidogenic Proteins and Secreted β-Amyloid Peptide Cultured cells from the commercial HUVECs supply were examined immunocytochemically to verify their cellular profile. Cells grown 4 - 10 days in vitro were mostly polygonal and sometimes fusiform in shape, had short somal protrusions and processes. All these cells expressed CD31, a marker of the vascular endothelia (Figure 1(A), Figure 1(D) and Figure 1(G)). In double immunofluorescence, virtually all CD31 expressing cells also co-labeled with antibodies to APP (Figures 1(A)-(C)), BACE1 (Figures 1(D)-(F)) [20] [32] and PS1-NTF (Figures 1(G)-(I)) [41]. To check the expression of amyloidogenic proteins biochemically, HUVECs lysates were blotted in parallel with human cortical lysates (available from our recent study). In comparison, HUVECs lysates contained less abundant holo-APP relative to brain samples, with the blotted protein bands migrated at a slightly heavier molecular weight position in the former (Figure 1(J)). Mature BACE1 (~70 Kd) and APP β-CTF (~14 Kd) were present in cortical and HUVECs lysates at similar migration position (Figure 1(J)). The signal of β-actin was comparable between the two assayed samples, whereas that of the neuronal protein β-tubulin was much more intense in the cortical relative to endothelial lysates (Figure 1(J)). To check Aβ secretion by HUVECs, the amounts of Aβ40 (considered to be most abundant Aβ species in CAA) in culture media were assayed with an ELISA kit. In several preliminary trials with media collected following 3 days of culture, Aβ40 levels were found to be around or slightly above the detecting threshold of kit. Subsequently, a batch of culture was carried out such that inpidual wells were maintained to different time points without change/resupply of culture medium. Thus, media from the wells with cells cultured for 2, 4, 6, 8, and 10 days were assayed for the levels of Aβ40. The amount of Aβ40 in the medium showed a trend of accumulation in the groups cultured from 2 - 10 days (Figure 1(K)). 3.2. Cholesterol Exposure Affected the Viability of Cultured HUVECs Pilot studies were carried out with cholesterol at titered concentrations from 200 down to 6.25 to mg/dL and duration of culture time from 24 - 48 hours, which indicated that the 6.25 mg/dL treatment had no noticeable effect relative to the 0 mg/dL group, whereas 200 mg/dL treatment caused massive cell death. Therefore, formal experiments (3 separate sets) were designed to determine the effect of 48 hour cholesterol coincubation at 0, 12.5, 25, 50 and 100 mg/dL concentrations, respectively. At the end of the treatments, we assessed cell morphology and the amount of cells microscopically as well as their viability using the MTT assay. Cell pallet lysates and culture media were assayed biochemically to quantify amyloidogenic protein and Aβ secretion. On microscopic examination with phase-contrast imaging, cells in the groups cultured with cholesterol at 0, 12.5 and 25 mg/dL were well attached to the coverslippers and showed healthy-looking morphology and comparable density (Figures 2(A)-(C) and Figures 2(A’)-(C’)). In contrast, cells in the 50 and 100 mg/dL groups appeared reduced or largely lost, with small-sized round profiles (likely cellular remains or debris) seen over microscopic field and only a few fusiform cells distinguishable (Figure 2(D) and Figure 2(D’), Figure 2(E) and Figure 2(E’)). In parallel, cell viability was affected by cholesterol coincubation. Thus, relative to control group (0 mg/dL), the MTT scores of the cholesterol treatment groups showed a dose-dependent reduction (P < 0.0001, F = 32.6, df = 4, 10, one-way ANOVA). Posthoc tests (Bonferroni’s Multiple Comparison Test) indicated a significant difference between the 0 mg/dL and all other dosage groups; between the 12.5 relative to the 50 and 100 mg/dL groups, and between the 25 relative to the 50 and 100 mg/dL groups (Figure 2(F)). 3.3. Cholesterol Exposure Affected the Amyloidogenic Capability of Cultured HUVECs Overall, the amounts of immunoblotted proteins were under the levels of detection (or lost) in the cell lysates from the group that was treated with 100 mg/dL cholesterol, therefore this group was not included in densitometric quantification (Figure 3(A)). Comparing to the control group (0 mg/dL), levels of holo-APP proteins were increased in the 12.5 and 25 mg/dL groups but reduced in the 50 mg/dL group (P < 0.0001, F = 66.0, df = 3, 8), with post-hoc tests indicating difference for the 12.5 vs. the 25 and the 50 mg/dL groups, and for the 25 vs. 50 mg/dL groups (Figure 3(B)). BACE1 protein levels were elevated in the 12.5 and 50 mg/dL groups relative to G. Z. Huang et al. 27 Figure 1. Expression of amyloidogenic proteins and Aβ production in cultured human umbilical vein endothelium cells (HUVECs). Panels (A)-(I) shows HUVECs grown in vitro 8 days after an initial expansion from the cell line, appearing in fusiform and polygonal profiles, and they express the endothelial signature protein CD31 (A) (D) (G). These cells also exhibit immunofluorescence for β-amyloid precursor protein (APP) (with the C-terminal antibody CT20), β-secretase 1 (BACE1) and presenilin-1 (PS1) (with the PS1-NTF antibody ab14). In cell lysates (J), the holo-APP proteins in HUVECs migrate at a slightly heavier molecular position relative to the counterpart immunoblotted in human brain cortical lysates. BACE1 and APP β-cleavage C-terminal fragment (APP β-CTF) are present in both the brain and HUVECs lysates (J). Note that the amount of β-actin is comparable between the two types of lysate, while the quantity of β-tubulin is minimal in the HUVECs lysates relative to human cortical samples used as assay control (J). The amount of Aβ40 in the media assayed by ELISA increases with extension of the culture days (d) (K). Insert in (K) shows a linear relationship between Aβ concentrations and light absorbance in ELISA. Scale bar = 100 μm in (A), applying to (B)-(I). G. Z. Huang et al. 28 Figure 2. Morphological characterization of the effect of cholesterol exposure on viability of cultured human umbilical vein endothelium cells (HUVECs). HUVECs are cultured using the same batch of cells for 4 days, followed by 48 hour treatment of cholesterol at 0 ((A), (A’)), 12.5 ((B), (B’)), 25 ((C), (C’)), 50 ((D), (D’)) and 100 ((E), (E)’) mg/dL concentrations and examined at the end of treatments under phase contrast microscope. The cells in the 0 - 25 mg/dL groups are healthy-looking and grown over the coverslippers ((A)-(C), (A’)-(C’)). Cells appear greatly reduced or essentially lost in the groups with 50 and 100 mg/dL cholesterol treatment. ((D), (E), (D’), (E’)). Panel (F) shows the relative viability scores with data normalized to the 0 mg/dL control group (defined as 100%), based on 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) fluogenic assay. There is a dose-dependent decline in cell viability (P < 0.0001, F = 32.6, df = 4, 10, one-way ANOVA), with posthoc test showing significant difference (* P < 0.05) in the 25 to 100 mg/dL groups relative to control. control (P < 0.0001, F = 17.3, df = 3, 8), showing difference for the 0 vs. the 25 mg/dL and for the 12.5 and 25 vs. the 50 mg/dL groups by post-hoc test (Figure 3(C)). Levels of APP β-CTF tended to increase in the 12.5 and 24 mg/dL groups but reduce in the 50 mg/dL group relative to control (P < 0.0001, F = 57.1, df = 3, 8), with significant difference between each pair of comparing dosage groups except for the 0 vs. 12.5 mg/dL groups (Figure 3(D)). Furthermore, APP β-site cleavage enzymatic activity exhibited a trend of increase in the 12.5 and 50 mg/dL groups and a reduction in the 100 mg/dL group relative to control (P < 0.0001, F = 32.8, df = 4, 10). Post-hoc test reported significant difference for the 0 vs. 25 mg/dL, the 12.5 vs. the 25 and 100 mg/dL groups, the 25 vs. 50 and 100 mg/dL and the 50 vs. 100 mg/dL groups (Figure 3(E)). Finally, Aβ40 levels in the media were elevated among the cholesterol treatment groups relative to control except for the 100 mg/dL group (P < 0.0001, F = 31.0, df = 4, 10). There existed significant difference for the control relative to the 25 and 50 mg/dL groups, for the 12.5 relative to the 25 and the 50 mg/dL groups, for the 25 relative to the 50 and 100 mg/dL groups by post-hoc test (Figure 3(F)). 4. Discussion 4.1. Vascular Endothelial Aβ Genesis and Its Potential Pharmacological Implications Earlier studies have demonstrated that several vascular endothelial cell lines, including the HUVECs and brain microvascular endothelial cells (BMECs), express the substrate and enzymes for Aβ production [35]-[39]. These findings are supported by in vivo observations from mouse and human brain preparations lately [40]-[42]. We have shown that APP, β-CTF (C99), BACE1 and PS1 protein products are detectable in primary culture of isolated human arterial endothelial cells [41]. According to an early report, APP770 is the major variant of the APP isoforms expressed at the protein levels by vascular endothelial cells, whereas neurons largely express APP695 G. Z. Huang et al. 29 Figure 3. Biochemical characterization of the effect of cholesterol exposure on amyloidogenic capability of cultured human umbilical vein endothelium cells (HUVECs). Panel (A) shows immunoblot images of cell lysate proteins from one set of experiments, documenting a loss of proteins in the 100 mg/dL group, with the amounts of APP, BACE1 and APP β-CTF, but not PS1-NTF, noticeably increased in the 12.5 and 25 mg/dL groups relative to control. Quantitatively, levels APP (B), BACE1 (C) and APP β-CTF (D) are significantly elevated (* ) or tended to increase (# ) in these two groups as compared to control. β-site APP cleavage (BACE1) enzymatic activity of the cell lysate is higher in the 12.5 - 50 mg/dL groups relative to control, with the 25 mg/dL group exhibiting statistical significance (E). Aβ40 levels in the media are elevated in the 12.5 - 50 mg/dL groups relative to control (F). * P < 0.05 relative to control. [37]. Consistent with this notion, the holo-APP proteins immunoblotted in primary human endothelial cultures migrate at a slightly heavier molecular weight position relative to its counterpart from brain lysates [41]. Importantly, data from humans and transgenic mouse models of AD suggest that Aβ overproduction in cerebral vascular endothelia, potentially mediated by excessive BACE1 activity, may contribute to the development of CAA [40]-[42], especially at the early pathogenic stage [41]. Here we have verified morphologically and biochemically that cultured HUVECs possess the full set of biochemical machinery of amyloidogenesis. Thus, these cells exhibit immunolabeling for APP and BACE1, as well as PS1 that serves the catalytic subunit of the γ-secretase complex [22] [55]. As with primary human vascular endothelial cultures [41], the holo-APP proteins immunoblotted with HUVECs lysates are slightly heavier than brain-derived APP, suggesting that the former likely represents the APP770 variant [37]. Moreover, Cultured HUVECs can secrete Aβ peptides, which accumulate in the media with time, as indicated by the increase of G. Z. Huang et al. 30 Aβ40 levels assessed by ELISA over different periods of cultivation. The finding that vascular endothelial cells produce Aβ may have some implications especially with regard to the design of drugs to antagonize brain amyloid pathogenesis. Attenuating Aβ overproduction by BACE1 and γ-secretase inhibitors/modulators has been explored as an pharmacological option for AD or cerebral amyloid pathology, although clinical trials on a number of γ-secretase inhibitors have failed to establish therapeutic benefits so far [56]. BACE1 inhibitors appear to be promising given that they block the initial step of the amyloidogenic pathway, and can reduce the levels of Aβ as well as APP β-CTF, the latter is also considered to be neurotoxic [18] [21] [57]-[60]. However, emerging evidence also raises concern as to whether inhibition of neuronal BACE1 may cause serious side-effects by disrupting the neurobiological role of the enzyme [61]. It is of interest to note that initially developed BACE1 inhibitors are generally not advanced into clinical trials because they are incapable of passing across the Blood Brain Barrier (BBB). In this regard, targeting endothelial Aβ genesis with brain impermeable BACE1 inhibitors may be not only pharmacologically feasible but turn out to be a blessing strategy [42]. Hypothetically, such inhibitors could act against the early rise of Aβ in vascular wall, protect BBB integrity and have less neuronal toxic effect, and therefore, may serve as preventive agents. 4.2. Cholesterol as an Inductive Factor for Vascular Aβ Overproduction Cholesterol has been long considered a risk factor for vascular dementia and AD according to epidemiologic data [43]-[53]. Cholesterol is transported between tissues/cells via apolipoprotein E and its interaction with its cell-surface receptors [46]. Inpiduals carrying the apolipoprotein E epsilon 4 allele (APOE-ε4) are shown to have an increased vulnerability to AD and dementia [48] [51]. These carries are associated with hypercholesterolemia [43] [48] [53] and increased prevalence and severity of cerebral amyloid pathology, especially CAA [2] [11] [52] [62]. Statins, which lower blood cholesterol levels, appear to be beneficial for preventing AD and dementia [49] [63]. Given vascular endothelia being positioned at the first front to potential circulatory stress factors; it is of interest to identify such factors that might affect the amyloidogenic activity in these cells. As elaborated above, cholesterol represents one of the candidate circulatory stress factors of interest to explore its influence on endothelial Aβ genesis. In the present study we find that cultured HUVECs respond to cholesterol exposure in a dose-dependent biphasic mode. At the lower doses tested in vitro, from approximately 12.5 to 25 mg/dL, cholesterol exposure potentiates amyloidogenesis in HUVECs by up-regulating APP and BACE1, but not PS1, expression. At higher concentrations, especially at 50 and 100 mg/dL, cholesterol exposure reduces the viability and causes death of the HUVECs. The cellular toxicity induced by high cholesterol exposure is associated with diminished expression of the amyloidogenic (reduce or loss of APP, BACE1 and β-CTF immunoblotting signal) and house-keeping proteins (loss of GADPH immunoblotting signal), and reduced Aβ production (lowering of Aβ levels in culture medium), in the HUVECs. These findings imply that elevated blood cholesterol could be a factor to potentiate amyloidogenesis in vascular endothelial cells. In clinical practise, hypercholesterolemia is often defined by blood total cholesterol concentration greater than 200 mg/dL (this cutoff may vary from hospital to hospital). In the current in vitro setting, cultured HUVECs appeared to be very sensitive to exogenous cholesterol challenge. Indeed, at concentrations much lower than 200 mg/dL, they already responded to cholesterol exposure quite robustly (from enhanced Aβ genesis and even to compromised whole cell viability). The dose-effect response observed in our in vitro experiments was progressive relative to the cholesterol concentrations tested. Notably, the low concentration treatments (12.5 mg/dL as well as 6.25 mg/dL tested in pilot studies) did not apparently impair cell morphology, viability or responses of protein (e.g., APP, BACE1 or β-actin) expression. Therefore, while our findings could (and should) not be truly correlated to hypercholesterolemia in a clinical term (perhaps as it is always for the case of in vitro experiments), the data clearly demonstrate a modulatory effect of cholesterol on amyloidogenesis in vascular endothelial cells. Translation of in vitro findings towards clinical relevance needs to be always cautious. Bearing an interpretative limitation in mind, the results obtained in this study are nonetheless relevant to the understanding of a potential pathophysiological role of hypercholesterolemia. 5. Conclusion In summary, cultured human umbilical vein endothelial cells express a full set of biochemical machinery for the production of β-amyloid peptides and are capable of secreting the peptides into culture medium. The amyloido- G. Z. Huang et al. 31 genic activity of these cells is enhanced in response to the exposure of cholesterol within a certain concentration range, while strong cholesterol challenge deteriorates their viability. These findings point to a possibility that hypercholesterolemia may serve as a circulatory factor to promote β-amyloid production in and release from vascular endothelial cells. Acknowledgements We thank Edward Koo, Huaibin Cai and Samuel E Gandy for providing the APP, BACE1 and PS1-NTF antibodies. This study was supported by National Natural Science Foundation of China (#81171160, #31371095) and approved by the Ethical Committee for biomedical research at Xiangya School of Medicine. The authors declare that the research was conducted in the absence of any commercial or financial relationships. References [1] Castellani, R.J., Smith, M.A., Perry, G. and Friedland, R.P. (2004) Cerebral Amyloid Angiopathy: Major Contributor or Decorative Response to Alzheimer’s Disease Pathogenesis. Neurobiology Aging, 25, 599-602. http://dx.doi.org/10.1016/j.neurobiolaging.2003.12.019 [2] Thal, D.R., Griffin, W.S., de Vos, R.A. and Ghebremedhin, E. (2008) Cerebral Amyloid Angiopathy and Its Relationship to Alzheimer’s Disease. Acta Neuropathologica, 115, 599-609. http://dx.doi.org/10.1007/s00401-008-0366-2 [3] Weller, R.O., Boche, D. and Nicoll, J.A. (2009) Microvasculature Changes and Cerebral Amyloid Angiopathy in Alzheimer’s Disease and Their Potential Impact on Therapy. Acta Neuropathologica, 118, 87-102. http://dx.doi.org/10.1007/s00401-009-0498-z [4] Honjo, K., Black, S.E. and Verhoeff, N.P. (2012) Alzheimer’s Disease, Cerebrovascular Disease, and the β-Amyloid Cascade. Canadian Journal of Neurological Sciences, 39, 712-728. http://dx.doi.org/10.1017/S0317167100015547 [5] Pfeifer, L.A., White, L.R., Ross, G.W., Petrovitch, H. and Launer, L.J. (2002) Cerebral Amyloid Angiopathy and Cognitive Function: The HAAS Autopsy Study. Neurology, 58, 1629-1634. http://dx.doi.org/10.1212/WNL.58.11.1629 [6] Jellinger, K.A. and Attems, J. (2003) Incidence of Cerebrovascular Lesions in Alzheimer’s Disease: A Postmortem Study. Acta Neuropathologica, 105, 14-17. [7] Keage, H.A., Carare, R.O., Friedland, R.P., Ince, P.G., Love, S., Nicoll, J.A., Wharton, S.B., Weller, R.O. and Brayne, C. (2009) Population Studies of Sporadic Cerebral Amyloid Angiopathy and Dementia: A Systematic Review. BMC Neurology, 9, 3. http://dx.doi.org/10.1186/1471-2377-9-3 [8] Arvanitakis, Z., Leurgans, S.E., Wang, Z., Wilson, R.S., Bennett, D.A. and Schneider, J.A. (2011) Cerebral Amyloid Angiopathy Pathology and Cognitive Domains in Older Persons. Annals of Neurology, 69, 320-327. http://dx.doi.org/10.1002/ana.22112 [9] Obrien, R.J. (2011) Vascular Dementia: Atherosclerosis, Cognition and Alzheimer’s Disease. Current Alzheimer Research, 8, 341-344. http://dx.doi.org/10.2174/156720511795745267 [10] Viswanathan, A. and Greenberg, S.M. (2011) Cerebral Amyloid Angiopathy in the Elderly. Annals of Neurology, 70, 871-880. http://dx.doi.org/10.1002/ana.22516 [11] Purrucker, J.C., Hund, E., Ringleb, P.A., Hartmann, C., Rohde, S., Schönland, S. and Steiner, T. (2013) Cerebral Amyloid Angiopathy—An Underdiagnosed Entity in Younger Adults with Lobar Intracerebral Hemorrhage? Amyloid, 20, 45-47. http://dx.doi.org/10.3109/13506129.2012.746937 [12] Charidimou, A., Gang, Q. and Werring, D.J. (2012) Sporadic Cerebral Amyloid Angiopathy Revisited: Recent Insights into Pathophysiology and Clinical Spectrum. Journal of Neurology, Neurosurgery, and Psychiatry, 83, 124-137. http://dx.doi.org/10.1136/jnnp-2011-301308 [13] Allen, N., Robinson, A.C., Snowden, J., Davidson, Y.S. and Mann, D.M. (2014) Patterns of Cerebral Amyloid Angiopathy Define Histopathological Phenotypes in Alzheimer’s Disease. Neuropathology and Applied Neurobiology, 40, 136-148. http://dx.doi.org/10.1111/nan.12070 [14] Geddes, J.W. and Cotman, C.W. (1991) Plasticity in Alzheimer’s Disease: Too Much or Not Enough? Neurobiology of Aging, 12, 330-333. http://dx.doi.org/10.1016/0197-4580(91)90011-8 [15] Arendt, T. (2001) Alzheimer’s Disease as a Disorder of Mechanisms Underlying Structural Brain Self-Organization. Neuroscience, 102, 723-765. http://dx.doi.org/10.1016/S0306-4522(00)00516-9 [16] Castellani, R.J., Lee, H.G., Zhu, X., Perry, G. and Smith, M.A. (2008) Alzheimer Disease Pathology as a Host Response. Journal of Neuropathology and Experimental Neurology, 67, 523-531. http://dx.doi.org/10.1097/NEN.0b013e318177eaf4 G. Z. Huang et al. 32 [17] Li, J.M., Liu, C., Hu, X., Cai, Y., Ma, C., Luo, X.G. and Yan, X.X. (2014) Inverse Correlation between Alzheimer’s Disease and Cancer: Implication for a Strong Impact of Regenerative Propensity on Neurodegeneration? BMC Neurology, 14, 211. http://dx.doi.org/10.1186/s12883-014-0211-2 [18] Yan, X.X., Ma, C., Gai, W.P., Cai, H. and Luo, X.G. (2014) Can BACE1 Inhibition Mitigate Early Axonal Pathology in Neurological Diseases? Journal of Alzheimer’s Disease, 38, 705-718 [19] Li, J.M., Cai, Y., Liu, F., Yang, L., Hu, X., Patrylo, P.R., Cai, H., Luo, X.G., Xiao, D. and Yan, X.X. (2015) Experimental Microembolism Induces Localized Neuritic Pathology in Guinea Pig Cerebrum. Oncotarget, 6, 10772-10785. http://dx.doi.org/10.18632/oncotarget.3599 [20] Laird, F.M., Cai, H., Savonenko, A.V., Farah, M.H., He, K., Melnikova, T., Wen, H., Chiang, H.C., Xu, G., Koliatsos, V.E., Borchelt, D.R., Price, D.L., Lee, H.K. and Wong, P.C. (2005) BACE1, a Major Determinant of Selective Vulnerability of the Brain to Amyloid-Beta Amyloidogenesis, Is Essential for Cognitive, Emotional, and Synaptic Functions. Journal of Neuroscience, 25, 11693-11709. http://dx.doi.org/10.1523/JNEUROSCI.2766-05.2005 [21] Vassar, R., Kovacs, D.M., Yan, R. and Wong, P.C. (2009) The Beta-Secretase Enzyme BACE in Health and Alzheimer’s Disease: Regulation, Cell Biology, Function, and Therapeutic Potential. Journal of Neuroscience, 29, 12787- 12794. http://dx.doi.org/10.1523/JNEUROSCI.3657-09.2009 [22] Liu, F., Xue, Z.Q., Deng, S.H., Kun, X., Luo, X.G., Patrylo, P.R., Rose, G.M., Cai, H., Struble, R.G., Cai, Y. and Yan, X.X. (2013) γ-Secretase Binding Sites in Aged and Alzheimer’s Disease Human Cerebrum: the Choroid Plexus as a Putative Origin of CSF Aβ. European Journal of Neuroscience, 37, 1714-1725. http://dx.doi.org/10.1111/ejn.12159 [23] Holsinger, R.M., McLean, C.A., Beyreuther, K., Masters, C.L. and Evin, G. (2002) Increased Expression of the Amyloid Precursor Beta-Secretase in Alzheimer’s Disease. Annals of Neurology, 51, 783-786. http://dx.doi.org/10.1002/ana.10208 [24] Tyler, S.J., Dawbarn, D., Wilcock, G.K. and Allen, S.J. (2002) Alpha- and Beta-Secretase: Profound Changes in Alzheimer’s Disease. Biochemical and Biophysical Research Communications, 299, 373-376. http://dx.doi.org/10.1016/S0006-291X(02)02635-9 [25] Fukumoto, H., Cheung, B.S., Hyman, B.T. and Irizarry, M.C. (2002) Beta-Secretase Protein and Activity are Increased in the Neocortex in Alzheimer Disease. Archives of Neurology, 59, 1381-1389. http://dx.doi.org/10.1001/archneur.59.9.1381 [26] Yang, L.B., Lindholm, K., Yan, R., Citron, M., Xia, W., Yang, X.L., Beach, T., Sue, L., Wong, P., Price, D., Li, R. and Shen, Y. (2003) Elevated Beta-Secretase Expression and Enzymatic Activity Detected in Sporadic Alzheimer Disease. Nature Medicine, 9, 3-4. http://dx.doi.org/10.1038/nm0103-3 [27] Li, R., Lindholm, K., Yang, L.B., Yue, X., Citron, M., Yan, R., Beach, T., Sue, L., Sabbagh, M., Cai, H., Wong, P., Price, D. and Shen, Y. (2004) Amyloid Beta Peptide Load Is Correlated with Increased Beta-Secretase Activity in Sporadic Alzheimer’s Disease Patients. Proceedings of the National Academy of Sciences of the United States of America, 101, 3632-3637. http://dx.doi.org/10.1073/pnas.0205689101 [28] Coulson, D.T., Beyer, N., Quinn, J.G., Brockbank, S., Hellemans, J., Irvine, G.B., Ravid, R. and Johnston, J.A. (2010) BACE1 mRNA Expression in Alzheimer’s Disease Postmortem Brain Tissue. Journal of Alzheimer’s Disease, 22, 1111-1122. [29] Cai, Y., Xiong, K., Zhang, X.M., Cai, H., Luo, X.G., Feng, J.C., Clough, R.W., Struble, R.G., Patrylo, P.R., Chu, Y., Kordower, J.H. and Yan, X.X. (2010) β-Secretase-1 Elevation in Aged Monkey and Alzheimer’s Disease Human Cerebral Cortex Occurs around the Vasculature in Partnership with Multisystem Axon Terminal Pathogenesis and β-Amyloid Accumulation. European Journal of Neuroscience, 32, 1223-1238. http://dx.doi.org/10.1111/j.1460-9568.2010.07376.x [30] Cheng, X., He, P., Lee, T., Yao, H., Li, R. and Shen, Y. (2014) High Activities of BACE1 in Brains with Mild Cognitive Impairment. American Journal of Pathology, 184, 141-147. http://dx.doi.org/10.1016/j.ajpath.2013.10.002 [31] Zhao, J., Fu, Y., Yasvoina, M., Shao, P., Hitt, B., O’Connor, T., Logan, S., Maus, E., Citron, M., Berry, R., Binder, L. and Vassar, R. (2007) Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 Levels Become Elevated in Neurons around Amyloid Plaques: Implications for Alzheimer’s Disease Pathogenesis. Journal of Neuroscience, 27, 3639-3649. http://dx.doi.org/10.1523/JNEUROSCI.4396-06.2007 [32] Zhang, X.M., Cai, Y., Cai, H., Xiong, K., Luo, X.G., Feng, J.C., Clough, R.W., Struble, R.G., Patrylo, P.R. and Yan, X.X. (2009) BACE1 Elevation in Transgenic Mouse Models of Alzheimer’s Disease Is Associated with Synaptic/Axonal Pathology and Amyloidogenesis: Implications for Neuritic Plaque Development. European Journal of Neuroscience, 30, 2271-2283. http://dx.doi.org/10.1111/j.1460-9568.2009.07017.x [33] Cai, Y., Zhang, X.M., Macklin, L.N., Cai, H., Luo, X.G., Oddo, S., Laferla, F.M., Struble, R.G., Rose, G.M., Patrylo, P.R. and Yan, X.X. (2012) BACE1 Elevation Is Involved in Amyloid Plaque Development in the Triple Transgenic Model of Alzheimer’s Disease: Differential Aβ Antibody Labeling of Early-Onset Axon Terminal Pathology. Neurotoxicity Research, 21, 160-174. http://dx.doi.org/10.1007/s12640-011-9256-9 G. Z. Huang et al. 33 [34] Kandalepas, P.C., Sadleir, K.R., Eimer, W.A., Zhao, J., Nicholson, D.A. and Vassar, R. (2013) The Alzheimer’s β-Secretase BACE1 Localizes to Normal Presynaptic Terminals and to Dystrophic Presynaptic Terminals Surrounding Amyloid Plaques. Acta Neuropathologica, 126, 329-352. http://dx.doi.org/10.1007/s00401-013-1152-3 [35] Simons, E.R., Marshall, D.C., Long, H.J., Otto, K., Billingslea, A., Tibbles, H., Wells, J., Eisenhauer, P., Fine, R.E., Cribbs, D.H., Davies, T.A. and Abraham, C.R. (1998) Blood Brain Barrier Endothelial Cells Express Candidate Amyloid Precursor Protein-Cleaving Secretases. Amyloid, 5, 153-162. http://dx.doi.org/10.3109/13506129809003841 [36] Austin, S.A., Santhanam, A.V. and Katusic, Z.S. (2010) Endothelial Nitric Oxide Modulates Expression and Processing of Amyloid Precursor Protein. Circulation Research, 107, 1498-502. http://dx.doi.org/10.1161/CIRCRESAHA.110.233080 [37] Kitazume, S., Tachida, Y., Kato, M., Yamaguchi, Y., Honda, T., Hashimoto, Y., Wada, Y., Saito, T., Iwata, N., Saido, T. and Taniguchi, N. (2010) Brain Endothelial Cells Produce Amyloid β from Amyloid Precursor Protein 770 and Preferentially Secrete the O-Glycosylated Form. The Journal of Biological Chemistry, 285, 40097-40103. http://dx.doi.org/10.1074/jbc.M110.144626 [38] Ma, J.F., Wang, H.M., Li, Q.Y., Zhang, Y., Pan, J., Qiang, Q., Xin, X.Y., Tang, H.D., Ding, J.Q. and Chen, S.D. (2010) Starvation Triggers Abeta42 Generation from Human Umbilical Vascular Endothelial Cells. FEBS Letters, 584, 3101- 3106. http://dx.doi.org/10.1016/j.febslet.2010.05.048 [39] Bulbarelli, A., Lonati, E., Brambilla, A., Orlando, A., Cazzaniga, E., Piazza, F., Ferrarese, C., Masserini, M. and Sancini, G. (2012) Aβ42 Production in Brain Capillary Endothelial Cells after Oxygen and Glucose Deprivation. Molecular and Cellular Neuroscience, 49, 415-422. http://dx.doi.org/10.1016/j.mcn.2012.01.007 [40] Cheng, X., He, P., Yao, H., Dong, Q., Li, R. and Shen, Y. (2014) Occludin Deficiency with BACE1 Elevation in Cerebral Amyloid Angiopathy. Neurology, 82, 1707-1715. http://dx.doi.org/10.1212/WNL.0000000000000403 [41] Xue, Z.Q., He, Z.W., Yu, J.J., Cai, Y., Qiu, W.Y., Pan, A., Gai, W.P., Cai, H., Luo, X.G., Ma, C. and Yan, X.X. (2015) Non-Neuronal and Neuronal BACE1 Elevation in Association with Angiopathic and Leptomeningeal β-Amyloid Deposition in the Human Brain. BMC Neurology, 15, 71. http://dx.doi.org/10.1186/s12883-015-0327-z [42] Devraj, K., Poznanovic, S., Spahn, C., Schwall, G., Harter, P.N., Mittelbronn, M., Antoniello, K., Paganetti. P., Muhs, A., Heilemann, M., Hawkins, R.A., Schrattenholz, A. and Liebner, S. (2015) BACE-1 Is Expressed in the Blood-Brain Barrier Endothelium and Is Upregulated in a Murine Model of Alzheimer’s Disease. Journal of Cerebral Blood Flow and Metabolism. http://dx.doi.org/10.1177/0271678x15606463 [43] Utermann, G., Kindermann, I., Kaffarnik, H. and Steinmetz, A. (1984) Apolipoprotein E Phenotypes and Hyperlipidemia. Human Genetics, 65, 232-236. http://dx.doi.org/10.1007/BF00286508 [44] Li, G., Shofer, J.B., Kukull, W.A., Peskind, E.R., Tsuang, D.W., Breitner, J.C., McCormick, W., Bowen, J.D., Teri, L., Schellenberg, G.D. and Larson, E.B. (2005) Serum Cholesterol and Risk of Alzheimer Disease: A Community-Based Cohort Study. Neurology, 65, 1045-1050. http://dx.doi.org/10.1212/01.wnl.0000178989.87072.11 [45] Anstey, K.J., Lipnicki, D.M. and Low, L.F. (2008) Cholesterol as a Risk Factor for Dementia and Cognitive Decline: A Systematic Review of Prospective Studies with Meta-Analysis. American Journal of Geriatric Psychiatry, 16, 343- 354. http://dx.doi.org/10.1097/01.JGP.0000310778.20870.ae [46] Mahley, R.W., Huang, Y. and Weisgraber, K.H. (2006) Putting Cholesterol in Its Place: ApoE and Reverse Cholesterol Transport. Journal of Clinical Investigation, 116, 1226-1229. http://dx.doi.org/10.1172/JCI28632 [47] Solomon, A., Kivipelto, M., Wolozin, B., Zhou, J. and Whitmer, R.A. (2009) Midlife Serum Cholesterol and Increased Risk of Alzheimer’s and Vascular Dementia Three Decades Later. Dementia and Geriatric Cognitive Disorder, 28, 75- 80. http://dx.doi.org/10.1159/000231980 [48] Leduc, V., Jasmin-Bélanger, S. and Poirier, J. (2010) APOE and Cholesterol Homeostasis in Alzheimer’s Disease. Trends in Molecular Medicine, 16, 469-477. http://dx.doi.org/10.1016/j.molmed.2010.07.008 [49] Shepardson, N.E., Shankar, G.M. and Selkoe, D.J. (2011) Cholesterol Level and Statin Use in Alzheimer Disease: II. Review of Human Trials and Recommendations. Archives of Neurology, 68, 1385-1392. http://dx.doi.org/10.1001/archneurol.2011.242 [50] Tolppanen, A.M., Solomon, A., Soininen, H. and Kivipelto, M. (2012) Midlife Vascular Risk Factors and Alzheimer’s Disease: Evidence from Epidemiological Studies. Journal of Alzheimer’s Disease, 32, 531-540. [51] Sadigh-Eteghad, S., Talebi, M. and Farhoudi, M. (2012) Association of Apolipoprotein E Epsilon 4 Allele with Sporadic Late Onset Alzheimer’s Disease. A Meta-Analysis. Neurosciences (Riyadh), 17, 321-326. [52] Silva, T., Teixeira, J., Remião, F. and Borges, F. (2013) Alzheimer’s Disease, Cholesterol, and Statins: The Junctions of Important Metabolic Pathways. Angewandte Chemie International Edition in English, 52, 1110-1121. http://dx.doi.org/10.1002/anie.201204964 [53] Reed, B., Villeneuve, S., Mack, W., Decarli, C., Chui, H.C. and Jagust, W. (2014) Associations between Serum Cholesterol Levels and Cerebral Amyloidosis. JAMA Neurology, 71, 195-200. G. Z. Huang et al. 34 http://dx.doi.org/10.1001/jamaneurol.2013.5390 [54] Maulik, M., Westaway, D., Jhamandas, J.H. and Kar, S. (2013) Role of Cholesterol in APP Metabolism and Its Significance in Alzheimer’s Disease Pathogenesis. Molecular Neurobiology, 47, 37-63. http://dx.doi.org/10.1007/s12035-012-8337-y [55] Yan, X.X., Li, T., Rominger, C.M., Prakash, S.R., Wong, P.C., Olson, R.E., Zaczek, R. and Li, Y.W. (2004) Binding Sites of Gamma-Secretase Inhibitors in Rodent Brain: Distribution, Postnatal Development, and Effect of Deafferentation. The Journal of Neuroscience, 24, 2942-2952. http://dx.doi.org/10.1523/JNEUROSCI.0092-04.2004 [56] De Strooper, B. and Chávez Gutiérrez, L. (2015) Learning by Failing: Ideas and Concepts to Tackle γ-Secretases in Alzheimer’s Disease and beyond. Annual Review of Pharmacology and Toxicology, 55, 419-437. http://dx.doi.org/10.1146/annurev-pharmtox-010814-124309 [57] Kim, H.S., Park, C.H., Cha, S.H., Lee, J.H., Lee, S., Kim, Y., Rah, J.C., Jeong, S.J. and Suh, Y.H. (2000) Carboxyl-Terminal Fragment of Alzheimer’s APP Destabilizes Calcium Homeostasis and Renders Neuronal Cells Vulnerable to Excitotoxicity. The FASEB Journal, 14, 1508-1517. http://dx.doi.org/10.1096/fj.14.11.1508 [58] Saganich, M.J., Schroeder, B.E., Galvan, V., Bredesen, D.E., Koo, E.H. and Heinemann, S.F. (2006) Deficits in Synaptic Transmission and Learning in Amyloid Precursor Protein (APP) Transgenic Mice Require C-Terminal Cleavage of APP. The Journal of Neuroscience, 26, 13428-13436. http://dx.doi.org/10.1523/JNEUROSCI.4180-06.2006 [59] Lauritzen, I., Pardossi-Piquard, R., Bauer, C., Brigham, E., Abraham, J.D., Ranaldi, S., Fraser, P., St-George-Hyslop, P., Le Thuc, O., Espin, V., Chami, L., Dunys, J. and Checler, F. (2012) The β-Secretase-Derived C-Terminal Fragment of βAPP, C99, but Not Aβ, Is a Key Contributor to Early Intraneuronal Lesions in Triple-Transgenic Mouse Hippocampus. The Journal of Neuroscience, 32, 16243-16255. http://dx.doi.org/10.1523/JNEUROSCI.2775-12.2012 [60] Jiang, Y., Rigoglioso, A., Peterhoff, C.M., Pawlik, M., Sato, Y., Bleiwas, C., Stavrides, P., Smiley, J.F., Ginsberg, S.D., Mathews, P.M., Levy, E. and Nixon, R.A. (2016) Partial BACE1 Reduction in a Down Syndrome Mouse Model Blocks Alzheimer-Related Endosomal Anomalies and Cholinergic Neurodegeneration: Role of APP-CTF. Neurobiology of Aging, 39, 90-98. http://dx.doi.org/10.1016/j.neurobiolaging.2015.11.013 [61] Barão, S., Moechars, D., Lichtenthaler, S.F. and De Strooper, B. (2016) BACE1 Physiological Functions May Limit Its Use as Therapeutic Target for Alzheimer’s Disease. Trends in Neurosciences, 39, 158-269. http://dx.doi.org/10.1016/j.tins.2016.01.003 [62] Nicoll, J.A. and McCarron, M.O. (2001) APOE Gene Polymorphism as a Risk Factor for Cerebral Amyloid Angiopathy-Related Hemorrhage. Amyloid, 8, 51-55. [63] Kurata, T., Kawai, H., Miyazaki, K., Kozuki, M., Morimoto, N., Ohta, Y., Ikeda, Y. and Abe, K. (2012) Statins Have Therapeutic Potential for the Treatment of Alzheimer’s Disease, Likely via Protection of the Neurovascular Unit in the AD Brain. Journal of the Neurological Sciences, 322, 59-63. http://dx.doi.org/10.1016/j.jns.2012.06.011Cholesterol Potentiates β-Amyloid Genesis in Cultured Human Umbilical VeinEnd…|in有
Cholesterol Potentiates β-Amyloid Genesis in Cultured Human Umbilical Vein Endothelial Cells Guozhi Huang1,2, Zhiqin Xue3, Xia Hu1, Lily Wan1, Jianming Li4, Yan Cai1, Peter R. Patrylo5, Xuegang Luo1, Aihua Pan1, Xiao-Xin Yan1,6* 1 Department of Anatomy and Neurobiology, Central South University Xiangya Medical School, Changsha, China 2 Department of Anatomy & Embryology, Loudi Vocational and Technical College, Loudi, China 3 Department of Anatomy, Xinjiang Medical University, Urumqi, China 4 Neuroscience Research Center, Changsha Medical University, Changsha, China 5 Departments of Physiology, Anatomy and the Center for Integrated Research in Cognitive and Neural Sciences, Southern Illinois University Carbondale, Carbondale, USA 6 Key Laboratory of Hunan Province in Neurodegenerative Disorders, Changsha, China Received 20 April 2016; accepted 3 June 2016; published 6 June 2016 Copyright © 2016 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Cerebral Amyloid Angiopathy (CAA) occurs commonly among the elderly and almost invariably in patients with Alzheimer’s Disease (AD). The β-amyloid peptides (Aβ) are produced via the amyloidogenic processing of β-Amyloid Precursor Protein (APP) by β-secretase-1 (BACE1) and γ- secretase. Vascular endothelial cells are lately shown to possess the molecular machinery of Aβ production, which might participate in the development of CAA. Hypercholesterolemia is considered a risk factor for AD, whereas less is known if cholesterol may modulate endothelial Aβ production. In the present study we verified the amyloidogenic capability of Human Umbilical Vein Endothelial Cells (HUVECs) in vitro and explored the effect of cholesterol exposure on their amyloidogenic potential. Cholesterol treatments at 12.5 and 25 mg/dL significantly elevated APP, BACE1 and APP β-CTF protein levels and β-site APP cleavage activity in cell lysates, and Aβ40 levels in culture medium. However, coincubation with cholesterol at 50 and 100 mg/dL attenuated the viability of the cultured cells and diminished their amyloidogenic capability. These findings suggest that high cholesterol exposure is stressful to vascular endothelial cells, and at a certain dosage range can promote an amyloidogenic response in these cells. * Corresponding author. G. Z. Huang et al. 24 Keywords Aging, Alzheimer’s Disease, Cholesterol, Neurodegeneration, Vascular Dementia 1. Introduction Cerebral amyloidosis is a pathological condition caused by the deposition of the β-amyloid peptides (Aβ) in the extracellular matrix and vascular wall in the central nervous system, referred to as parenchymal amyloid plaques and Cerebral Amyloid Angiopathy (CAA), respectively [1] [2]. Amyloid plaques and CAA are among the definitive neuropathological lesions of dementia of the Alzheimer’s Disease (AD) and vascular types [3]-[5], although both types of amyloidosis can be seen in the brain of non-demented elderly [6]-[13]. Along with Aβ deposits, parenchymal and vascular amyloid pathology may coexist with degenerative and reactive cellular changes, including neuronal and vascular cell loss, neuritic dystrophy and glial activation [14]-[19]. Neurons appear to be the major origin of soluble and insoluble Aβ products in the brain given that they are enriched with the amyloidogenic biochemical machinery including β-Amyloid Precursor Protein (APP), β-secretatse-1 (BACE1) and γ-secretase [20]-[22]. In particular, elevated BACE1 protein and enzymatic activity are reported in the human brains with amyloid neuropathology [22]-[30]. Specifically, enhanced BACE1 expression is identified microscopically in dystrophic axons around neuritic and angiopathic Aβ deposition [18] [31]-[34]. Vascular endothelial cells are initially shown to express the full set of amyloidogenic machinery in vitro, which has been lately demonstrated in vivo including in the human brain with CAA [35]-[42]. It is suggested that Aβ overproduction by vascular endothelial cells may be involved in the early stages of the development of CAA [40]-[42]. Therefore, it becomes important to identify circulatory factors that might potentiate endothelial amyloidogenesis. Hypercholesterolemia has been considered a risk factor for AD and vascular dementia for a long time [43]-[53]. However, it remains less clear if cholesterol may directly influence the amyloidogenic activity in vascular endothelial cells [54]. In the present study, we used Human Umbilical Vein Endothelial Cells (HUVECs) as a model to explore a modulatory effect of cholesterol on endothelial amyloidogenic protein expression and Aβ secretion. Cultured HUVECs exhibited a biphasic response to cholesterol treatments, with low dose exposure potentiated Aβ genesis while high dose exposure resulted in cell death and diminished Aβ production. 2. Materials and Methods 2.1. Cell Culture and Cholesterol Treatment HUVECs were obtained commercially (Catalog #C0035C, Life Technologies Corporation). Cell supplies were re-suspended with DMEM/F12 containing 10% fetal bovine serum, to a beginning density approximately 5 × 106 cells/mL. Cells were cultured at 37˚C in a humidified atmosphere containing 95% air and 5% CO2, in six-well Corning-Costar plates (Catalog #CLS3516; Life Technologies Corporation). Each well was loaded with 2 ml of medium and a glass coverslip coated with polylysine, with the medium changed twice a week. Initial experiments were carried out to verify the cultured cells and optimize the biochemical assays. Cholesterol treatments to HUVECs were initiated on day 4 of culture for each round of experiments for duration of 48 hours. A total of 3 experiments were carried out separately in different times. For each experiment, media containing 0, 12.5, 25, 50 and 100 mg/dL of cholesterol (Catalog #C8667, Sigma-Aldrich, St. Louis, MO, USA) were added in duplicate into wells loaded with the same amount of cells from the same original batch of HUVECs expansion. Media were collected following 48 hours of culture to assess Aβ40 concentrations, with the corresponding groups of cultured cells harvested for morphological study and assessment of related proteins and APP β-cleavage activity. 2.2. Immunocytochemistry and Microscopic Examination HUVECs grown on coverslippers were fixed in situ with cold 4% paraformaldehyde for 20 minutes and stored frozen until immunofluorescent processing. Coverslippers were thawed to room temperature prior to staining, and incubated in PBS containing 5% donkey serum for 30 minutes, followed by reaction with a pair of primary G. Z. Huang et al. 25 antibodies raised in different species overnight at 4˚C, including 1) mouse anti-CD31 (endothelial marker) [41] and rabbit anti-APP C-terminal (CT20); 2) mouse anti-CD31 and rabbit anti-BACE1 [20] [32]; 3) mouse antiCD31 and rabbit anti-presilinin-1 (PS1) N-terminal (ab14) [41]. Following several rinses with PBS, the coverslippers were further incubated at room temperature for a 2 hour with Alexa Fluor® 488 donkey anti-rabbit and Alexa Fluor® 594 conjugated donkey anti-mouse IgGs (1:200, Invitrogen, Carlsbad, CA, USA). Coverslippers were finally counter-stained with bisbenzimide (Hoechst 33,342, 1:50,000, Catalog #B2261, Sigma-Aldrich), washed thoroughly, and mounted with anti-fading medium. Immunofluorescence was examined on an Olympus fluorescent BX53 microscope equipped with a digital imaging system (CellSens Standard, Olympus Corporation, Japan). 2.3. Western Blot HUVECs pellets (×4, w⁄v) were homogenized by sonication in T-PER extraction buffer (Pierce, Rockford, IL, USA) containing protease inhibitors (Roche, Indianapolis, IN, USA). The lysates were centrifuged at 15,000 g, with supernatants collected and protein concentrations determined by DC protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Human cortical lysates available from a previous study (Xue et al., 2015) were used as an assay control to cross-validate the protein profile of the HUVECs relative to brain tissue. Fifty microgram protein was run on each lane in 10% or 18% (for APP β-CTF) SDS-polyacrylamide electrophoresis gels. Separated proteins were electrotransferred to Trans-Blot pure nitrocellulose membranes (Bio-Rad Lab.), which were immunoblotted with antibodies to APP (22C11), APP β-CTF (AHP538), BACE1, PS1-NTF (ab14), β-tubulin-III, β-actin at pre-optimized working dilutions with agitation for 12 hours. The membranes were further reacted with horseradish peroxidase (HRP)-conjugated goat anti-rabbit or anti-mouse IgGs (1:20,000; Bio-Rad Laboratories), treated with the ECL Plus Western Blotting Detection Kit, and exposed against Kodak X-OMAT Blue autoradiography films that were developed in a darkroom subsequently. 2.4. APP β-Site Cleavage (BACE1) Activity Assay APP β-site cleavage activity (representing BACE1 enzymatic activity) in HUVECs was measured in duplicate in 96-well transparent flat-bottomed plates. Samples were homogenized on ice as described above for Western blot, and assayed for enzymatic activity using a commercial kit (Calbiochem, La Jolla, CA, USA, Catalog #565785) according to manufacturer’s instructions. The fluorescent signal was captured in a Bio-Rad microplate reader (PR 3100 TSC). 2.5. Enzyme-Linked Immunosorbent Assay for Aβ in Culture Medium Aβ levels in cell culture media were obtained to estimate the amount of secreted peptides following cultivation for different time periods or treatments of cholesterol at different concentrations. Media were assayed in duplicate by solid sandwich enzyme-linked immunosorbent assay (ELISA) to assess Aβ40 levels using commercial kits (Novex® KHB3482 for Aβ40) following the manufacturer’s instruction (Life Technologies Corporation). Signals were captured in the Bio-Rad microplate reader, with the Aβ concentrations (pM/ml) calculated according to the standard curve generated from the readouts of the synthesized peptides supplied along with the kit. 2.6. MTT Cell Viability Assay The viability of HUVECs was assayed at the end of cholesterol treatment experiments using a commercial fluogenic 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) kit following the manufacturer’s instruction (Abnova, Catalog # KA1606, Taipei City, Taiwan). Fluorescent signal was measured in the Bio-Rad microplate reader, with cell viability calculated for comparing culture groups. 2.7. Quantification, Statistical Analysis and Figure Preparation Densitometric (Western blot, digitalized using NIH-Image J) and numeric data (light absorbent signals of APP β-site cleavage enzymatic activity, Aβ ELISA and MTT fluogenic activity) were normalized to either the internal standard or the control group. Means were calculated for comparing groups, and analyzed statistically using one-way ANOVA with Bonferroni’s multiple comparison tests (Prism GraphPad, San Diego, CA). The significant level of difference was set at P < 0.05. Figures were assembled with Photoshop 7.0, with brightness/ G. Z. Huang et al. 26 contract adjusted as needed. 3. Results 3.1. HUVECs Expressed Amyloidogenic Proteins and Secreted β-Amyloid Peptide Cultured cells from the commercial HUVECs supply were examined immunocytochemically to verify their cellular profile. Cells grown 4 - 10 days in vitro were mostly polygonal and sometimes fusiform in shape, had short somal protrusions and processes. All these cells expressed CD31, a marker of the vascular endothelia (Figure 1(A), Figure 1(D) and Figure 1(G)). In double immunofluorescence, virtually all CD31 expressing cells also co-labeled with antibodies to APP (Figures 1(A)-(C)), BACE1 (Figures 1(D)-(F)) [20] [32] and PS1-NTF (Figures 1(G)-(I)) [41]. To check the expression of amyloidogenic proteins biochemically, HUVECs lysates were blotted in parallel with human cortical lysates (available from our recent study). In comparison, HUVECs lysates contained less abundant holo-APP relative to brain samples, with the blotted protein bands migrated at a slightly heavier molecular weight position in the former (Figure 1(J)). Mature BACE1 (~70 Kd) and APP β-CTF (~14 Kd) were present in cortical and HUVECs lysates at similar migration position (Figure 1(J)). The signal of β-actin was comparable between the two assayed samples, whereas that of the neuronal protein β-tubulin was much more intense in the cortical relative to endothelial lysates (Figure 1(J)). To check Aβ secretion by HUVECs, the amounts of Aβ40 (considered to be most abundant Aβ species in CAA) in culture media were assayed with an ELISA kit. In several preliminary trials with media collected following 3 days of culture, Aβ40 levels were found to be around or slightly above the detecting threshold of kit. Subsequently, a batch of culture was carried out such that inpidual wells were maintained to different time points without change/resupply of culture medium. Thus, media from the wells with cells cultured for 2, 4, 6, 8, and 10 days were assayed for the levels of Aβ40. The amount of Aβ40 in the medium showed a trend of accumulation in the groups cultured from 2 - 10 days (Figure 1(K)). 3.2. Cholesterol Exposure Affected the Viability of Cultured HUVECs Pilot studies were carried out with cholesterol at titered concentrations from 200 down to 6.25 to mg/dL and duration of culture time from 24 - 48 hours, which indicated that the 6.25 mg/dL treatment had no noticeable effect relative to the 0 mg/dL group, whereas 200 mg/dL treatment caused massive cell death. Therefore, formal experiments (3 separate sets) were designed to determine the effect of 48 hour cholesterol coincubation at 0, 12.5, 25, 50 and 100 mg/dL concentrations, respectively. At the end of the treatments, we assessed cell morphology and the amount of cells microscopically as well as their viability using the MTT assay. Cell pallet lysates and culture media were assayed biochemically to quantify amyloidogenic protein and Aβ secretion. On microscopic examination with phase-contrast imaging, cells in the groups cultured with cholesterol at 0, 12.5 and 25 mg/dL were well attached to the coverslippers and showed healthy-looking morphology and comparable density (Figures 2(A)-(C) and Figures 2(A’)-(C’)). In contrast, cells in the 50 and 100 mg/dL groups appeared reduced or largely lost, with small-sized round profiles (likely cellular remains or debris) seen over microscopic field and only a few fusiform cells distinguishable (Figure 2(D) and Figure 2(D’), Figure 2(E) and Figure 2(E’)). In parallel, cell viability was affected by cholesterol coincubation. Thus, relative to control group (0 mg/dL), the MTT scores of the cholesterol treatment groups showed a dose-dependent reduction (P < 0.0001, F = 32.6, df = 4, 10, one-way ANOVA). Posthoc tests (Bonferroni’s Multiple Comparison Test) indicated a significant difference between the 0 mg/dL and all other dosage groups; between the 12.5 relative to the 50 and 100 mg/dL groups, and between the 25 relative to the 50 and 100 mg/dL groups (Figure 2(F)). 3.3. Cholesterol Exposure Affected the Amyloidogenic Capability of Cultured HUVECs Overall, the amounts of immunoblotted proteins were under the levels of detection (or lost) in the cell lysates from the group that was treated with 100 mg/dL cholesterol, therefore this group was not included in densitometric quantification (Figure 3(A)). Comparing to the control group (0 mg/dL), levels of holo-APP proteins were increased in the 12.5 and 25 mg/dL groups but reduced in the 50 mg/dL group (P < 0.0001, F = 66.0, df = 3, 8), with post-hoc tests indicating difference for the 12.5 vs. the 25 and the 50 mg/dL groups, and for the 25 vs. 50 mg/dL groups (Figure 3(B)). BACE1 protein levels were elevated in the 12.5 and 50 mg/dL groups relative to G. Z. Huang et al. 27 Figure 1. Expression of amyloidogenic proteins and Aβ production in cultured human umbilical vein endothelium cells (HUVECs). Panels (A)-(I) shows HUVECs grown in vitro 8 days after an initial expansion from the cell line, appearing in fusiform and polygonal profiles, and they express the endothelial signature protein CD31 (A) (D) (G). These cells also exhibit immunofluorescence for β-amyloid precursor protein (APP) (with the C-terminal antibody CT20), β-secretase 1 (BACE1) and presenilin-1 (PS1) (with the PS1-NTF antibody ab14). In cell lysates (J), the holo-APP proteins in HUVECs migrate at a slightly heavier molecular position relative to the counterpart immunoblotted in human brain cortical lysates. BACE1 and APP β-cleavage C-terminal fragment (APP β-CTF) are present in both the brain and HUVECs lysates (J). Note that the amount of β-actin is comparable between the two types of lysate, while the quantity of β-tubulin is minimal in the HUVECs lysates relative to human cortical samples used as assay control (J). The amount of Aβ40 in the media assayed by ELISA increases with extension of the culture days (d) (K). Insert in (K) shows a linear relationship between Aβ concentrations and light absorbance in ELISA. Scale bar = 100 μm in (A), applying to (B)-(I). G. Z. Huang et al. 28 Figure 2. Morphological characterization of the effect of cholesterol exposure on viability of cultured human umbilical vein endothelium cells (HUVECs). HUVECs are cultured using the same batch of cells for 4 days, followed by 48 hour treatment of cholesterol at 0 ((A), (A’)), 12.5 ((B), (B’)), 25 ((C), (C’)), 50 ((D), (D’)) and 100 ((E), (E)’) mg/dL concentrations and examined at the end of treatments under phase contrast microscope. The cells in the 0 - 25 mg/dL groups are healthy-looking and grown over the coverslippers ((A)-(C), (A’)-(C’)). Cells appear greatly reduced or essentially lost in the groups with 50 and 100 mg/dL cholesterol treatment. ((D), (E), (D’), (E’)). Panel (F) shows the relative viability scores with data normalized to the 0 mg/dL control group (defined as 100%), based on 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) fluogenic assay. There is a dose-dependent decline in cell viability (P < 0.0001, F = 32.6, df = 4, 10, one-way ANOVA), with posthoc test showing significant difference (* P < 0.05) in the 25 to 100 mg/dL groups relative to control. control (P < 0.0001, F = 17.3, df = 3, 8), showing difference for the 0 vs. the 25 mg/dL and for the 12.5 and 25 vs. the 50 mg/dL groups by post-hoc test (Figure 3(C)). Levels of APP β-CTF tended to increase in the 12.5 and 24 mg/dL groups but reduce in the 50 mg/dL group relative to control (P < 0.0001, F = 57.1, df = 3, 8), with significant difference between each pair of comparing dosage groups except for the 0 vs. 12.5 mg/dL groups (Figure 3(D)). Furthermore, APP β-site cleavage enzymatic activity exhibited a trend of increase in the 12.5 and 50 mg/dL groups and a reduction in the 100 mg/dL group relative to control (P < 0.0001, F = 32.8, df = 4, 10). Post-hoc test reported significant difference for the 0 vs. 25 mg/dL, the 12.5 vs. the 25 and 100 mg/dL groups, the 25 vs. 50 and 100 mg/dL and the 50 vs. 100 mg/dL groups (Figure 3(E)). Finally, Aβ40 levels in the media were elevated among the cholesterol treatment groups relative to control except for the 100 mg/dL group (P < 0.0001, F = 31.0, df = 4, 10). There existed significant difference for the control relative to the 25 and 50 mg/dL groups, for the 12.5 relative to the 25 and the 50 mg/dL groups, for the 25 relative to the 50 and 100 mg/dL groups by post-hoc test (Figure 3(F)). 4. Discussion 4.1. Vascular Endothelial Aβ Genesis and Its Potential Pharmacological Implications Earlier studies have demonstrated that several vascular endothelial cell lines, including the HUVECs and brain microvascular endothelial cells (BMECs), express the substrate and enzymes for Aβ production [35]-[39]. These findings are supported by in vivo observations from mouse and human brain preparations lately [40]-[42]. We have shown that APP, β-CTF (C99), BACE1 and PS1 protein products are detectable in primary culture of isolated human arterial endothelial cells [41]. According to an early report, APP770 is the major variant of the APP isoforms expressed at the protein levels by vascular endothelial cells, whereas neurons largely express APP695 G. Z. Huang et al. 29 Figure 3. Biochemical characterization of the effect of cholesterol exposure on amyloidogenic capability of cultured human umbilical vein endothelium cells (HUVECs). Panel (A) shows immunoblot images of cell lysate proteins from one set of experiments, documenting a loss of proteins in the 100 mg/dL group, with the amounts of APP, BACE1 and APP β-CTF, but not PS1-NTF, noticeably increased in the 12.5 and 25 mg/dL groups relative to control. Quantitatively, levels APP (B), BACE1 (C) and APP β-CTF (D) are significantly elevated (* ) or tended to increase (# ) in these two groups as compared to control. β-site APP cleavage (BACE1) enzymatic activity of the cell lysate is higher in the 12.5 - 50 mg/dL groups relative to control, with the 25 mg/dL group exhibiting statistical significance (E). Aβ40 levels in the media are elevated in the 12.5 - 50 mg/dL groups relative to control (F). * P < 0.05 relative to control. [37]. Consistent with this notion, the holo-APP proteins immunoblotted in primary human endothelial cultures migrate at a slightly heavier molecular weight position relative to its counterpart from brain lysates [41]. Importantly, data from humans and transgenic mouse models of AD suggest that Aβ overproduction in cerebral vascular endothelia, potentially mediated by excessive BACE1 activity, may contribute to the development of CAA [40]-[42], especially at the early pathogenic stage [41]. Here we have verified morphologically and biochemically that cultured HUVECs possess the full set of biochemical machinery of amyloidogenesis. Thus, these cells exhibit immunolabeling for APP and BACE1, as well as PS1 that serves the catalytic subunit of the γ-secretase complex [22] [55]. As with primary human vascular endothelial cultures [41], the holo-APP proteins immunoblotted with HUVECs lysates are slightly heavier than brain-derived APP, suggesting that the former likely represents the APP770 variant [37]. Moreover, Cultured HUVECs can secrete Aβ peptides, which accumulate in the media with time, as indicated by the increase of G. Z. Huang et al. 30 Aβ40 levels assessed by ELISA over different periods of cultivation. The finding that vascular endothelial cells produce Aβ may have some implications especially with regard to the design of drugs to antagonize brain amyloid pathogenesis. Attenuating Aβ overproduction by BACE1 and γ-secretase inhibitors/modulators has been explored as an pharmacological option for AD or cerebral amyloid pathology, although clinical trials on a number of γ-secretase inhibitors have failed to establish therapeutic benefits so far [56]. BACE1 inhibitors appear to be promising given that they block the initial step of the amyloidogenic pathway, and can reduce the levels of Aβ as well as APP β-CTF, the latter is also considered to be neurotoxic [18] [21] [57]-[60]. However, emerging evidence also raises concern as to whether inhibition of neuronal BACE1 may cause serious side-effects by disrupting the neurobiological role of the enzyme [61]. It is of interest to note that initially developed BACE1 inhibitors are generally not advanced into clinical trials because they are incapable of passing across the Blood Brain Barrier (BBB). In this regard, targeting endothelial Aβ genesis with brain impermeable BACE1 inhibitors may be not only pharmacologically feasible but turn out to be a blessing strategy [42]. Hypothetically, such inhibitors could act against the early rise of Aβ in vascular wall, protect BBB integrity and have less neuronal toxic effect, and therefore, may serve as preventive agents. 4.2. Cholesterol as an Inductive Factor for Vascular Aβ Overproduction Cholesterol has been long considered a risk factor for vascular dementia and AD according to epidemiologic data [43]-[53]. Cholesterol is transported between tissues/cells via apolipoprotein E and its interaction with its cell-surface receptors [46]. Inpiduals carrying the apolipoprotein E epsilon 4 allele (APOE-ε4) are shown to have an increased vulnerability to AD and dementia [48] [51]. These carries are associated with hypercholesterolemia [43] [48] [53] and increased prevalence and severity of cerebral amyloid pathology, especially CAA [2] [11] [52] [62]. Statins, which lower blood cholesterol levels, appear to be beneficial for preventing AD and dementia [49] [63]. Given vascular endothelia being positioned at the first front to potential circulatory stress factors; it is of interest to identify such factors that might affect the amyloidogenic activity in these cells. As elaborated above, cholesterol represents one of the candidate circulatory stress factors of interest to explore its influence on endothelial Aβ genesis. In the present study we find that cultured HUVECs respond to cholesterol exposure in a dose-dependent biphasic mode. At the lower doses tested in vitro, from approximately 12.5 to 25 mg/dL, cholesterol exposure potentiates amyloidogenesis in HUVECs by up-regulating APP and BACE1, but not PS1, expression. At higher concentrations, especially at 50 and 100 mg/dL, cholesterol exposure reduces the viability and causes death of the HUVECs. The cellular toxicity induced by high cholesterol exposure is associated with diminished expression of the amyloidogenic (reduce or loss of APP, BACE1 and β-CTF immunoblotting signal) and house-keeping proteins (loss of GADPH immunoblotting signal), and reduced Aβ production (lowering of Aβ levels in culture medium), in the HUVECs. These findings imply that elevated blood cholesterol could be a factor to potentiate amyloidogenesis in vascular endothelial cells. In clinical practise, hypercholesterolemia is often defined by blood total cholesterol concentration greater than 200 mg/dL (this cutoff may vary from hospital to hospital). In the current in vitro setting, cultured HUVECs appeared to be very sensitive to exogenous cholesterol challenge. Indeed, at concentrations much lower than 200 mg/dL, they already responded to cholesterol exposure quite robustly (from enhanced Aβ genesis and even to compromised whole cell viability). The dose-effect response observed in our in vitro experiments was progressive relative to the cholesterol concentrations tested. Notably, the low concentration treatments (12.5 mg/dL as well as 6.25 mg/dL tested in pilot studies) did not apparently impair cell morphology, viability or responses of protein (e.g., APP, BACE1 or β-actin) expression. Therefore, while our findings could (and should) not be truly correlated to hypercholesterolemia in a clinical term (perhaps as it is always for the case of in vitro experiments), the data clearly demonstrate a modulatory effect of cholesterol on amyloidogenesis in vascular endothelial cells. Translation of in vitro findings towards clinical relevance needs to be always cautious. Bearing an interpretative limitation in mind, the results obtained in this study are nonetheless relevant to the understanding of a potential pathophysiological role of hypercholesterolemia. 5. Conclusion In summary, cultured human umbilical vein endothelial cells express a full set of biochemical machinery for the production of β-amyloid peptides and are capable of secreting the peptides into culture medium. The amyloido- G. Z. Huang et al. 31 genic activity of these cells is enhanced in response to the exposure of cholesterol within a certain concentration range, while strong cholesterol challenge deteriorates their viability. These findings point to a possibility that hypercholesterolemia may serve as a circulatory factor to promote β-amyloid production in and release from vascular endothelial cells. Acknowledgements We thank Edward Koo, Huaibin Cai and Samuel E Gandy for providing the APP, BACE1 and PS1-NTF antibodies. This study was supported by National Natural Science Foundation of China (#81171160, #31371095) and approved by the Ethical Committee for biomedical research at Xiangya School of Medicine. The authors declare that the research was conducted in the absence of any commercial or financial relationships. References [1] Castellani, R.J., Smith, M.A., Perry, G. and Friedland, R.P. (2004) Cerebral Amyloid Angiopathy: Major Contributor or Decorative Response to Alzheimer’s Disease Pathogenesis. Neurobiology Aging, 25, 599-602. http://dx.doi.org/10.1016/j.neurobiolaging.2003.12.019 [2] Thal, D.R., Griffin, W.S., de Vos, R.A. and Ghebremedhin, E. (2008) Cerebral Amyloid Angiopathy and Its Relationship to Alzheimer’s Disease. Acta Neuropathologica, 115, 599-609. http://dx.doi.org/10.1007/s00401-008-0366-2 [3] Weller, R.O., Boche, D. and Nicoll, J.A. (2009) Microvasculature Changes and Cerebral Amyloid Angiopathy in Alzheimer’s Disease and Their Potential Impact on Therapy. Acta Neuropathologica, 118, 87-102. http://dx.doi.org/10.1007/s00401-009-0498-z [4] Honjo, K., Black, S.E. and Verhoeff, N.P. (2012) Alzheimer’s Disease, Cerebrovascular Disease, and the β-Amyloid Cascade. Canadian Journal of Neurological Sciences, 39, 712-728. http://dx.doi.org/10.1017/S0317167100015547 [5] Pfeifer, L.A., White, L.R., Ross, G.W., Petrovitch, H. and Launer, L.J. (2002) Cerebral Amyloid Angiopathy and Cognitive Function: The HAAS Autopsy Study. Neurology, 58, 1629-1634. http://dx.doi.org/10.1212/WNL.58.11.1629 [6] Jellinger, K.A. and Attems, J. (2003) Incidence of Cerebrovascular Lesions in Alzheimer’s Disease: A Postmortem Study. Acta Neuropathologica, 105, 14-17. [7] Keage, H.A., Carare, R.O., Friedland, R.P., Ince, P.G., Love, S., Nicoll, J.A., Wharton, S.B., Weller, R.O. and Brayne, C. (2009) Population Studies of Sporadic Cerebral Amyloid Angiopathy and Dementia: A Systematic Review. BMC Neurology, 9, 3. http://dx.doi.org/10.1186/1471-2377-9-3 [8] Arvanitakis, Z., Leurgans, S.E., Wang, Z., Wilson, R.S., Bennett, D.A. and Schneider, J.A. (2011) Cerebral Amyloid Angiopathy Pathology and Cognitive Domains in Older Persons. Annals of Neurology, 69, 320-327. http://dx.doi.org/10.1002/ana.22112 [9] Obrien, R.J. (2011) Vascular Dementia: Atherosclerosis, Cognition and Alzheimer’s Disease. Current Alzheimer Research, 8, 341-344. http://dx.doi.org/10.2174/156720511795745267 [10] Viswanathan, A. and Greenberg, S.M. (2011) Cerebral Amyloid Angiopathy in the Elderly. Annals of Neurology, 70, 871-880. http://dx.doi.org/10.1002/ana.22516 [11] Purrucker, J.C., Hund, E., Ringleb, P.A., Hartmann, C., Rohde, S., Schönland, S. and Steiner, T. (2013) Cerebral Amyloid Angiopathy—An Underdiagnosed Entity in Younger Adults with Lobar Intracerebral Hemorrhage? Amyloid, 20, 45-47. http://dx.doi.org/10.3109/13506129.2012.746937 [12] Charidimou, A., Gang, Q. and Werring, D.J. (2012) Sporadic Cerebral Amyloid Angiopathy Revisited: Recent Insights into Pathophysiology and Clinical Spectrum. Journal of Neurology, Neurosurgery, and Psychiatry, 83, 124-137. http://dx.doi.org/10.1136/jnnp-2011-301308 [13] Allen, N., Robinson, A.C., Snowden, J., Davidson, Y.S. and Mann, D.M. (2014) Patterns of Cerebral Amyloid Angiopathy Define Histopathological Phenotypes in Alzheimer’s Disease. Neuropathology and Applied Neurobiology, 40, 136-148. http://dx.doi.org/10.1111/nan.12070 [14] Geddes, J.W. and Cotman, C.W. (1991) Plasticity in Alzheimer’s Disease: Too Much or Not Enough? Neurobiology of Aging, 12, 330-333. http://dx.doi.org/10.1016/0197-4580(91)90011-8 [15] Arendt, T. (2001) Alzheimer’s Disease as a Disorder of Mechanisms Underlying Structural Brain Self-Organization. Neuroscience, 102, 723-765. http://dx.doi.org/10.1016/S0306-4522(00)00516-9 [16] Castellani, R.J., Lee, H.G., Zhu, X., Perry, G. and Smith, M.A. (2008) Alzheimer Disease Pathology as a Host Response. Journal of Neuropathology and Experimental Neurology, 67, 523-531. http://dx.doi.org/10.1097/NEN.0b013e318177eaf4 G. Z. Huang et al. 32 [17] Li, J.M., Liu, C., Hu, X., Cai, Y., Ma, C., Luo, X.G. and Yan, X.X. (2014) Inverse Correlation between Alzheimer’s Disease and Cancer: Implication for a Strong Impact of Regenerative Propensity on Neurodegeneration? BMC Neurology, 14, 211. http://dx.doi.org/10.1186/s12883-014-0211-2 [18] Yan, X.X., Ma, C., Gai, W.P., Cai, H. and Luo, X.G. (2014) Can BACE1 Inhibition Mitigate Early Axonal Pathology in Neurological Diseases? Journal of Alzheimer’s Disease, 38, 705-718 [19] Li, J.M., Cai, Y., Liu, F., Yang, L., Hu, X., Patrylo, P.R., Cai, H., Luo, X.G., Xiao, D. and Yan, X.X. (2015) Experimental Microembolism Induces Localized Neuritic Pathology in Guinea Pig Cerebrum. Oncotarget, 6, 10772-10785. http://dx.doi.org/10.18632/oncotarget.3599 [20] Laird, F.M., Cai, H., Savonenko, A.V., Farah, M.H., He, K., Melnikova, T., Wen, H., Chiang, H.C., Xu, G., Koliatsos, V.E., Borchelt, D.R., Price, D.L., Lee, H.K. and Wong, P.C. (2005) BACE1, a Major Determinant of Selective Vulnerability of the Brain to Amyloid-Beta Amyloidogenesis, Is Essential for Cognitive, Emotional, and Synaptic Functions. Journal of Neuroscience, 25, 11693-11709. http://dx.doi.org/10.1523/JNEUROSCI.2766-05.2005 [21] Vassar, R., Kovacs, D.M., Yan, R. and Wong, P.C. (2009) The Beta-Secretase Enzyme BACE in Health and Alzheimer’s Disease: Regulation, Cell Biology, Function, and Therapeutic Potential. Journal of Neuroscience, 29, 12787- 12794. http://dx.doi.org/10.1523/JNEUROSCI.3657-09.2009 [22] Liu, F., Xue, Z.Q., Deng, S.H., Kun, X., Luo, X.G., Patrylo, P.R., Rose, G.M., Cai, H., Struble, R.G., Cai, Y. and Yan, X.X. (2013) γ-Secretase Binding Sites in Aged and Alzheimer’s Disease Human Cerebrum: the Choroid Plexus as a Putative Origin of CSF Aβ. European Journal of Neuroscience, 37, 1714-1725. http://dx.doi.org/10.1111/ejn.12159 [23] Holsinger, R.M., McLean, C.A., Beyreuther, K., Masters, C.L. and Evin, G. (2002) Increased Expression of the Amyloid Precursor Beta-Secretase in Alzheimer’s Disease. Annals of Neurology, 51, 783-786. http://dx.doi.org/10.1002/ana.10208 [24] Tyler, S.J., Dawbarn, D., Wilcock, G.K. and Allen, S.J. (2002) Alpha- and Beta-Secretase: Profound Changes in Alzheimer’s Disease. Biochemical and Biophysical Research Communications, 299, 373-376. http://dx.doi.org/10.1016/S0006-291X(02)02635-9 [25] Fukumoto, H., Cheung, B.S., Hyman, B.T. and Irizarry, M.C. (2002) Beta-Secretase Protein and Activity are Increased in the Neocortex in Alzheimer Disease. Archives of Neurology, 59, 1381-1389. http://dx.doi.org/10.1001/archneur.59.9.1381 [26] Yang, L.B., Lindholm, K., Yan, R., Citron, M., Xia, W., Yang, X.L., Beach, T., Sue, L., Wong, P., Price, D., Li, R. and Shen, Y. (2003) Elevated Beta-Secretase Expression and Enzymatic Activity Detected in Sporadic Alzheimer Disease. Nature Medicine, 9, 3-4. http://dx.doi.org/10.1038/nm0103-3 [27] Li, R., Lindholm, K., Yang, L.B., Yue, X., Citron, M., Yan, R., Beach, T., Sue, L., Sabbagh, M., Cai, H., Wong, P., Price, D. and Shen, Y. (2004) Amyloid Beta Peptide Load Is Correlated with Increased Beta-Secretase Activity in Sporadic Alzheimer’s Disease Patients. Proceedings of the National Academy of Sciences of the United States of America, 101, 3632-3637. http://dx.doi.org/10.1073/pnas.0205689101 [28] Coulson, D.T., Beyer, N., Quinn, J.G., Brockbank, S., Hellemans, J., Irvine, G.B., Ravid, R. and Johnston, J.A. (2010) BACE1 mRNA Expression in Alzheimer’s Disease Postmortem Brain Tissue. Journal of Alzheimer’s Disease, 22, 1111-1122. [29] Cai, Y., Xiong, K., Zhang, X.M., Cai, H., Luo, X.G., Feng, J.C., Clough, R.W., Struble, R.G., Patrylo, P.R., Chu, Y., Kordower, J.H. and Yan, X.X. (2010) β-Secretase-1 Elevation in Aged Monkey and Alzheimer’s Disease Human Cerebral Cortex Occurs around the Vasculature in Partnership with Multisystem Axon Terminal Pathogenesis and β-Amyloid Accumulation. European Journal of Neuroscience, 32, 1223-1238. http://dx.doi.org/10.1111/j.1460-9568.2010.07376.x [30] Cheng, X., He, P., Lee, T., Yao, H., Li, R. and Shen, Y. (2014) High Activities of BACE1 in Brains with Mild Cognitive Impairment. American Journal of Pathology, 184, 141-147. http://dx.doi.org/10.1016/j.ajpath.2013.10.002 [31] Zhao, J., Fu, Y., Yasvoina, M., Shao, P., Hitt, B., O’Connor, T., Logan, S., Maus, E., Citron, M., Berry, R., Binder, L. and Vassar, R. (2007) Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 Levels Become Elevated in Neurons around Amyloid Plaques: Implications for Alzheimer’s Disease Pathogenesis. Journal of Neuroscience, 27, 3639-3649. http://dx.doi.org/10.1523/JNEUROSCI.4396-06.2007 [32] Zhang, X.M., Cai, Y., Cai, H., Xiong, K., Luo, X.G., Feng, J.C., Clough, R.W., Struble, R.G., Patrylo, P.R. and Yan, X.X. (2009) BACE1 Elevation in Transgenic Mouse Models of Alzheimer’s Disease Is Associated with Synaptic/Axonal Pathology and Amyloidogenesis: Implications for Neuritic Plaque Development. European Journal of Neuroscience, 30, 2271-2283. http://dx.doi.org/10.1111/j.1460-9568.2009.07017.x [33] Cai, Y., Zhang, X.M., Macklin, L.N., Cai, H., Luo, X.G., Oddo, S., Laferla, F.M., Struble, R.G., Rose, G.M., Patrylo, P.R. and Yan, X.X. (2012) BACE1 Elevation Is Involved in Amyloid Plaque Development in the Triple Transgenic Model of Alzheimer’s Disease: Differential Aβ Antibody Labeling of Early-Onset Axon Terminal Pathology. Neurotoxicity Research, 21, 160-174. http://dx.doi.org/10.1007/s12640-011-9256-9 G. Z. Huang et al. 33 [34] Kandalepas, P.C., Sadleir, K.R., Eimer, W.A., Zhao, J., Nicholson, D.A. and Vassar, R. (2013) The Alzheimer’s β-Secretase BACE1 Localizes to Normal Presynaptic Terminals and to Dystrophic Presynaptic Terminals Surrounding Amyloid Plaques. Acta Neuropathologica, 126, 329-352. http://dx.doi.org/10.1007/s00401-013-1152-3 [35] Simons, E.R., Marshall, D.C., Long, H.J., Otto, K., Billingslea, A., Tibbles, H., Wells, J., Eisenhauer, P., Fine, R.E., Cribbs, D.H., Davies, T.A. and Abraham, C.R. (1998) Blood Brain Barrier Endothelial Cells Express Candidate Amyloid Precursor Protein-Cleaving Secretases. Amyloid, 5, 153-162. http://dx.doi.org/10.3109/13506129809003841 [36] Austin, S.A., Santhanam, A.V. and Katusic, Z.S. (2010) Endothelial Nitric Oxide Modulates Expression and Processing of Amyloid Precursor Protein. Circulation Research, 107, 1498-502. http://dx.doi.org/10.1161/CIRCRESAHA.110.233080 [37] Kitazume, S., Tachida, Y., Kato, M., Yamaguchi, Y., Honda, T., Hashimoto, Y., Wada, Y., Saito, T., Iwata, N., Saido, T. and Taniguchi, N. (2010) Brain Endothelial Cells Produce Amyloid β from Amyloid Precursor Protein 770 and Preferentially Secrete the O-Glycosylated Form. The Journal of Biological Chemistry, 285, 40097-40103. http://dx.doi.org/10.1074/jbc.M110.144626 [38] Ma, J.F., Wang, H.M., Li, Q.Y., Zhang, Y., Pan, J., Qiang, Q., Xin, X.Y., Tang, H.D., Ding, J.Q. and Chen, S.D. (2010) Starvation Triggers Abeta42 Generation from Human Umbilical Vascular Endothelial Cells. FEBS Letters, 584, 3101- 3106. http://dx.doi.org/10.1016/j.febslet.2010.05.048 [39] Bulbarelli, A., Lonati, E., Brambilla, A., Orlando, A., Cazzaniga, E., Piazza, F., Ferrarese, C., Masserini, M. and Sancini, G. (2012) Aβ42 Production in Brain Capillary Endothelial Cells after Oxygen and Glucose Deprivation. Molecular and Cellular Neuroscience, 49, 415-422. http://dx.doi.org/10.1016/j.mcn.2012.01.007 [40] Cheng, X., He, P., Yao, H., Dong, Q., Li, R. and Shen, Y. (2014) Occludin Deficiency with BACE1 Elevation in Cerebral Amyloid Angiopathy. Neurology, 82, 1707-1715. http://dx.doi.org/10.1212/WNL.0000000000000403 [41] Xue, Z.Q., He, Z.W., Yu, J.J., Cai, Y., Qiu, W.Y., Pan, A., Gai, W.P., Cai, H., Luo, X.G., Ma, C. and Yan, X.X. (2015) Non-Neuronal and Neuronal BACE1 Elevation in Association with Angiopathic and Leptomeningeal β-Amyloid Deposition in the Human Brain. BMC Neurology, 15, 71. http://dx.doi.org/10.1186/s12883-015-0327-z [42] Devraj, K., Poznanovic, S., Spahn, C., Schwall, G., Harter, P.N., Mittelbronn, M., Antoniello, K., Paganetti. P., Muhs, A., Heilemann, M., Hawkins, R.A., Schrattenholz, A. and Liebner, S. (2015) BACE-1 Is Expressed in the Blood-Brain Barrier Endothelium and Is Upregulated in a Murine Model of Alzheimer’s Disease. Journal of Cerebral Blood Flow and Metabolism. http://dx.doi.org/10.1177/0271678x15606463 [43] Utermann, G., Kindermann, I., Kaffarnik, H. and Steinmetz, A. (1984) Apolipoprotein E Phenotypes and Hyperlipidemia. Human Genetics, 65, 232-236. http://dx.doi.org/10.1007/BF00286508 [44] Li, G., Shofer, J.B., Kukull, W.A., Peskind, E.R., Tsuang, D.W., Breitner, J.C., McCormick, W., Bowen, J.D., Teri, L., Schellenberg, G.D. and Larson, E.B. (2005) Serum Cholesterol and Risk of Alzheimer Disease: A Community-Based Cohort Study. Neurology, 65, 1045-1050. http://dx.doi.org/10.1212/01.wnl.0000178989.87072.11 [45] Anstey, K.J., Lipnicki, D.M. and Low, L.F. (2008) Cholesterol as a Risk Factor for Dementia and Cognitive Decline: A Systematic Review of Prospective Studies with Meta-Analysis. American Journal of Geriatric Psychiatry, 16, 343- 354. http://dx.doi.org/10.1097/01.JGP.0000310778.20870.ae [46] Mahley, R.W., Huang, Y. and Weisgraber, K.H. (2006) Putting Cholesterol in Its Place: ApoE and Reverse Cholesterol Transport. Journal of Clinical Investigation, 116, 1226-1229. http://dx.doi.org/10.1172/JCI28632 [47] Solomon, A., Kivipelto, M., Wolozin, B., Zhou, J. and Whitmer, R.A. (2009) Midlife Serum Cholesterol and Increased Risk of Alzheimer’s and Vascular Dementia Three Decades Later. Dementia and Geriatric Cognitive Disorder, 28, 75- 80. http://dx.doi.org/10.1159/000231980 [48] Leduc, V., Jasmin-Bélanger, S. and Poirier, J. (2010) APOE and Cholesterol Homeostasis in Alzheimer’s Disease. Trends in Molecular Medicine, 16, 469-477. http://dx.doi.org/10.1016/j.molmed.2010.07.008 [49] Shepardson, N.E., Shankar, G.M. and Selkoe, D.J. (2011) Cholesterol Level and Statin Use in Alzheimer Disease: II. Review of Human Trials and Recommendations. Archives of Neurology, 68, 1385-1392. http://dx.doi.org/10.1001/archneurol.2011.242 [50] Tolppanen, A.M., Solomon, A., Soininen, H. and Kivipelto, M. (2012) Midlife Vascular Risk Factors and Alzheimer’s Disease: Evidence from Epidemiological Studies. Journal of Alzheimer’s Disease, 32, 531-540. [51] Sadigh-Eteghad, S., Talebi, M. and Farhoudi, M. (2012) Association of Apolipoprotein E Epsilon 4 Allele with Sporadic Late Onset Alzheimer’s Disease. A Meta-Analysis. Neurosciences (Riyadh), 17, 321-326. [52] Silva, T., Teixeira, J., Remião, F. and Borges, F. (2013) Alzheimer’s Disease, Cholesterol, and Statins: The Junctions of Important Metabolic Pathways. Angewandte Chemie International Edition in English, 52, 1110-1121. http://dx.doi.org/10.1002/anie.201204964 [53] Reed, B., Villeneuve, S., Mack, W., Decarli, C., Chui, H.C. and Jagust, W. (2014) Associations between Serum Cholesterol Levels and Cerebral Amyloidosis. JAMA Neurology, 71, 195-200. G. Z. Huang et al. 34 http://dx.doi.org/10.1001/jamaneurol.2013.5390 [54] Maulik, M., Westaway, D., Jhamandas, J.H. and Kar, S. (2013) Role of Cholesterol in APP Metabolism and Its Significance in Alzheimer’s Disease Pathogenesis. Molecular Neurobiology, 47, 37-63. http://dx.doi.org/10.1007/s12035-012-8337-y [55] Yan, X.X., Li, T., Rominger, C.M., Prakash, S.R., Wong, P.C., Olson, R.E., Zaczek, R. and Li, Y.W. (2004) Binding Sites of Gamma-Secretase Inhibitors in Rodent Brain: Distribution, Postnatal Development, and Effect of Deafferentation. The Journal of Neuroscience, 24, 2942-2952. http://dx.doi.org/10.1523/JNEUROSCI.0092-04.2004 [56] De Strooper, B. and Chávez Gutiérrez, L. (2015) Learning by Failing: Ideas and Concepts to Tackle γ-Secretases in Alzheimer’s Disease and beyond. Annual Review of Pharmacology and Toxicology, 55, 419-437. http://dx.doi.org/10.1146/annurev-pharmtox-010814-124309 [57] Kim, H.S., Park, C.H., Cha, S.H., Lee, J.H., Lee, S., Kim, Y., Rah, J.C., Jeong, S.J. and Suh, Y.H. (2000) Carboxyl-Terminal Fragment of Alzheimer’s APP Destabilizes Calcium Homeostasis and Renders Neuronal Cells Vulnerable to Excitotoxicity. The FASEB Journal, 14, 1508-1517. http://dx.doi.org/10.1096/fj.14.11.1508 [58] Saganich, M.J., Schroeder, B.E., Galvan, V., Bredesen, D.E., Koo, E.H. and Heinemann, S.F. (2006) Deficits in Synaptic Transmission and Learning in Amyloid Precursor Protein (APP) Transgenic Mice Require C-Terminal Cleavage of APP. The Journal of Neuroscience, 26, 13428-13436. http://dx.doi.org/10.1523/JNEUROSCI.4180-06.2006 [59] Lauritzen, I., Pardossi-Piquard, R., Bauer, C., Brigham, E., Abraham, J.D., Ranaldi, S., Fraser, P., St-George-Hyslop, P., Le Thuc, O., Espin, V., Chami, L., Dunys, J. and Checler, F. (2012) The β-Secretase-Derived C-Terminal Fragment of βAPP, C99, but Not Aβ, Is a Key Contributor to Early Intraneuronal Lesions in Triple-Transgenic Mouse Hippocampus. The Journal of Neuroscience, 32, 16243-16255. http://dx.doi.org/10.1523/JNEUROSCI.2775-12.2012 [60] Jiang, Y., Rigoglioso, A., Peterhoff, C.M., Pawlik, M., Sato, Y., Bleiwas, C., Stavrides, P., Smiley, J.F., Ginsberg, S.D., Mathews, P.M., Levy, E. and Nixon, R.A. (2016) Partial BACE1 Reduction in a Down Syndrome Mouse Model Blocks Alzheimer-Related Endosomal Anomalies and Cholinergic Neurodegeneration: Role of APP-CTF. Neurobiology of Aging, 39, 90-98. http://dx.doi.org/10.1016/j.neurobiolaging.2015.11.013 [61] Barão, S., Moechars, D., Lichtenthaler, S.F. and De Strooper, B. (2016) BACE1 Physiological Functions May Limit Its Use as Therapeutic Target for Alzheimer’s Disease. Trends in Neurosciences, 39, 158-269. http://dx.doi.org/10.1016/j.tins.2016.01.003 [62] Nicoll, J.A. and McCarron, M.O. (2001) APOE Gene Polymorphism as a Risk Factor for Cerebral Amyloid Angiopathy-Related Hemorrhage. Amyloid, 8, 51-55. [63] Kurata, T., Kawai, H., Miyazaki, K., Kozuki, M., Morimoto, N., Ohta, Y., Ikeda, Y. and Abe, K. (2012) Statins Have Therapeutic Potential for the Treatment of Alzheimer’s Disease, Likely via Protection of the Neurovascular Unit in the AD Brain. Journal of the Neurological Sciences, 322, 59-63. http://dx.doi.org/10.1016/j.jns.2012.06.011