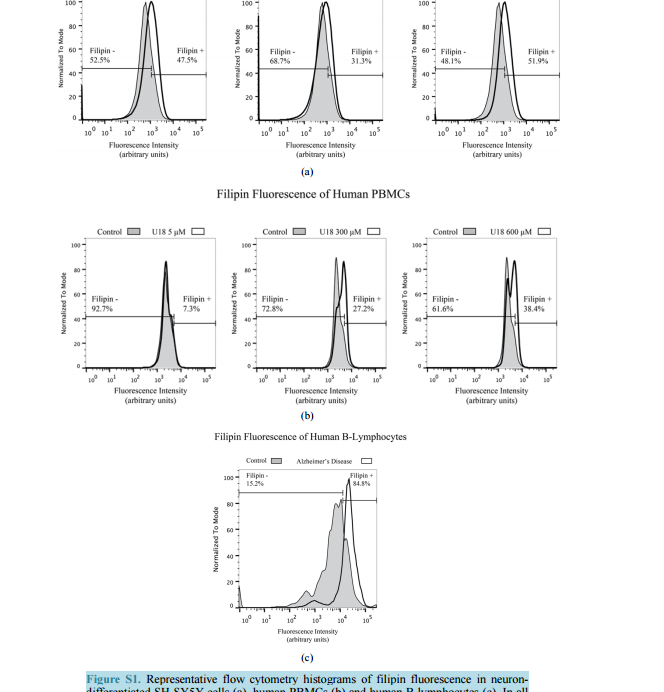
 Filipin Levels as Potential Predictors of Alzheimer’s Disease Risk Michael A. Castello, Kristy D. Howard, Arthur J. Castaneda, Salvador Soriano Division of Human Anatomy, Department of Pathology and Human Anatomy, Loma Linda University School of Medicine, Loma Linda, CA, USA Email: [email protected] Received 17 July 2014; revised 20 August 2014; accepted 28 August 2014 Copyright © 2014 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract To date, therapies to prevent or treat Alzheimer’s disease (AD) have largely focused on removing excess aggregation-prone amyloid peptide Aβ from the brain, an approach that has produced disappointing clinical outcomes. An alternative hypothesis proposes that Aβ production and aggregation is a symptom of a larger, systemic disease affecting the regulation of lipids, including cholesterol. In this scenario, lipid dysregulation would likely occur early in the disease process, making it an ideal target for predicting risk of mild cognitive impairment (MCI) to AD conversion. Here, we report that levels of filipin, a fluorescent polyene macrolide widely used as a diagnostic tool for diseases of lipid dysregulation, correlate with cellular damage caused by 27-hydroxycholesterol and with dementia status in human peripheral blood cells. These results provide strong preliminary data suggesting that filipin could be of use in the development of a quick and inexpensive method to measure the risk of AD conversion in patients with MCI, supplementing existing testing strategies that focus on the consequences of Aβ accumulation. Keywords Alzheimer’s Disease, Lipid Dysregulation, Cholesterol, Mild Cognitive Impairment, Filipin Levels 1. Introduction Alzheimer’s disease (AD) is the most common form of dementia in the United States, representing around eighty percent of all cases. As the country’s sixth leading cause of death, AD currently affects 5.4 million Americans and is projected to increase to 7.7 million over the next several decades [1]. Conclusive diagnosis of AD is made post mortem, when specific pathological hallmarks are visualized in the brain: extracellular senile plaques (SPs), comprised of the amyloid-beta peptide (Aβ), and neurofibrillary tangles (NFTs), made from abnormally phos- M. A. Castello et al. 138 phorylated tau protein. To date, AD research and therapy has largely focused on removing excess aggregationprone Aβ from the brain. However, while numerous drugs have successfully reduced the burden of Aβ in the brain by removing it or preventing its aggregation, they have failed to produce significant clinical improvements [2]. These lackluster outcomes have added weight to criticism from a growing number of scientists casting doubt on the validity of the amyloid hypothesis [3] [4]. One alternative that has been proposed by ourselves and others suggests that Aβ production and aggregation is a symptom of a larger, systemic disease that affects the exquisitely-regulated balance of lipids in the brain, including cholesterol [5]. Genome-wide association studies (GWAS), as well as epidemiological and biochemical evidence, place cholesterol regulation as a likely early pathogenic trigger of late onset Alzheimer’s disease, while biochemical and histochemical studies from human autopsy brains clearly show evidence for widespread brain cholesterol dysregulation in late onset AD [6]-[9]. Of particular interest is the presence of high levels of the side-chain oxidized form of cholesterol, 27-hydroxycholesterol (27-OHC), in the AD brain. Unlike cholesterol, 27-OHC can cross the blood brain barrier, providing a mechanism to account for the link between hypercholesterolemia and the risk of AD [10]. Studies using fibroblasts and peripheral blood cells from AD patients have added weight to this argument by supplying histological and biochemical evidence of lipid dysregulation taking place in those cells [11] [12]. At the same time, APP and Aβ production has been shown to be influenced by sterols including 27-OHC [13] [16]. If dysregulation begins at presymptomatic stages of AD, and its severity increases with disease progression, it might be possible to differentiate mild cognitive impairment (MCI) sufferers according to their risk of conversion into AD by monitoring dysregulation of cholesterol or other lipids in peripheral cells. Ideally, one would want an inexpensive test that can provide quick quantification of cholesterol dysregulation using a system amenable to high throughput approaches. Here, we report that filipin, a polyene macrolide widely used as a diagnostic tool for diseases of lipid dysregulation, such as Niemann-Pick type C disease (NPC), can be of potential use to develop such a test [17] [18]. We show that filipin fluorescence, as quantified by flow cytometry, can be measured in human peripheral blood mononuclear cells (PBMCs) and that its levels correlate with dementia status. These results provide strong support for larger clinical studies to test whether our filipin-based method can in fact predict the risk of AD conversion in MCI patients, an approach that would supplement existing strategies that focus on the consequences of Aβ accumulation. 2. Materials and Methods 2.1. Reagents U18666A (Sigma-Aldrich, St. Louis, MO) was stored in aliquots at -80˚C as an aqueous solution at a concentration of 4.7163 M (2 mg/ml). Working concentrations were prepared in culture medium immediately prior to experiments. 24-hydroxycholesterol (24-OHC) and 27-hydroxycholesterol (27-OHC) (Cayman Chemical, Ann Arbor, MI) were prepared as 10 mm stock solutions in 100% ethanol (Sigma-Aldrich, St. Louis, MO) and stored in aliquots at −80˚C. Working concentrations were prepared in culture medium immediately prior to experiments. Except where otherwise noted, cells were treated with media containing 5 mm U18666A, 10 µm 24-OHC, or 10 µm 27-OHC and incubated for 16 hours at 37˚C. 2.2. Cell Culture and Fluorescence Microscopy B-lymphocytes were obtained from Coriell cell repository (Camden, NJ; Table 1) and cultured in RPMI (Life Technologies, Carlsbad, CA) with 15% FBS (Atalanta Biologicals, Flowery Branch, GA) and 1% pen/strep (Life Technologies, Carlsbad, CA). According to information available from Coriell, AD patients were not known to be homozygous for ApoE4. SH-SY5Y human neuroblastoma cells (CRL-2266, American Type Culture Collection) were cultured in DMEM (Life Technologies, Carlsbad, CA) with 15% FBS and 1% pen/strep. For neuronal differentiation, cells were plated in 6-well plates at 400,000 cells/well (day 1). After 24 hrs (day 2), media was changed to DMEM with 3.75% FBS, 1% pen/strep, and 10 µm retinoic acid (RA; Sigma-Aldrich, St. Louis, MO). On day 5, media was discarded and replaced with fresh DMEM, again with 3.75% FBS, 1% pen/strep, and 10 µm RA. On day 6, M. A. Castello et al. 139 Table 1. Cells acquired from Coriell cell repository. According to the information available from Coriell, AD patients were not known to be homozygous for ApoE4. Cells Acquired from Coriell Cell Repository Catalog ID Group Age Cell Type AG08526 AD 61 B-Lymphocyte AG21157 AD 61 B-Lymphocyte AG06204 AD 67 B-Lymphocyte AG21159 AD 69 B-Lymphocyte AG08242 AD 72 B-Lymphocyte AG10986 Control 82 B-Lymphocyte AG10955 Control 89 B-Lymphocyte AG08897 Control 61 B-Lymphocyte media was changed to DMEM without FBS, and containing 10 µm RA with 50 ng/ml of brain-derived neurotrophic factor (BDNF; Sigma-Aldrich, St. Louis, MO). Differentiated cells were experimentally treated on day 8. Peripheral blood mononuclear cells (PBMCs) from adult donors were obtained from Leuko-pak leukocyte filters (Fenwal Laboratories, Lake Zurich, IL) and donated by the Blood Processing and Quality Control Lifestream (San Bernardino, CA). Ficoll-Hypaque (GE Healthcare, Pittsburgh, PA) and red blood cell lysis density gradient centrifugation were used for PBMC isolation exactly as described [19]. 2.3. Flow Cytometry For FACS analysis, cells were fixed in paraformaldehyde (PFA) at room temperature (RT; peripheral blood cells, 1% PFA 30 min at RT; B-lymphocytes, 4% PFA 1 hr at RT) and treated with filipin for 30 min at RT (peripheral blood cells, 50 μg/ml filipin; B-lymphocytes, 100 μg/ml filipin). Flow cytometry was performed using a MACSQuant Analyzer (Miltenyi Biotec, Bergisch Gladbach, Germany) with excitation at 350 nm. Mean fluorescence intensity was calculated with FlowJo software (TreeStar, Inc., Ashland, OR). Average percentage of filipin-positive to total number of cells was determined in FlowJo using flipin fluorescence in cells from control patients (B-lymphocytes) or vehicle-treated cells (SH-SY5Y and PBMCs) to determine cut-offs (as described in Figure S1). Statistical analysis of the percentages was performed in SPSS Statistics (version 22, IBM Corp.) using one-way ANOVA and Bonferroni post-hoc analysis. 3. Results To assess the impact of cholesterol dysregulation on cytotoxicity, we treated neuron-differentiated SH-SY5Y cells with side-chain oxidized cholesterol forms 24-OHC or 27-OHC, and measured extracellular lactate dehydrogenase (LDH) as an indicator of cell damage. In addition, we treated cells with U18666A, a compound that inhibits cholesterol synthesis and intracellular trafficking and leads to apoptosis in a variety of cell lines, including neurons, thus serving as positive control for cholesterol-associated cytotoxicity [20] [21]. As shown in Figure 1(a), treatment of cells with 27-OHC, but not 24-OHC, led to significantly higher levels of LDH release than in untreated control cells, in agreement with previous work from the Ghribi laboratory [15]. Note also that the cytotoxic impact of 27-OHC was comparable to that seen in cells treated with U18666. Next, we assessed the correlation between the cellular damage, as detected by LDH release, and levels of filipin fluorescence. Neuron-differentiated SH-SY5Y cells were treated as above with 24-OHC, 27-OHC, or U18666A, and fluorescence from filipin was measured by fluorescence-activated cell sorting (FACS). As seen in Figure 1(b), 27-OHC, but not 24-OHC, led to a statistically significant increase in filipin staining that was comparable to the impact of U18666A. This differential result indicates that, in our SH-SY5Y neuronal model, filipin fluorescence does not correlate with overall levels of oxidized cholesterol, because 24-OHC does not lead to significant changes in filipin signal. Rather, filipin fluorescence correlates with the cytotoxicity status that accompanies the changes in subcellular distribution of the lipids that it detects [22]. This correlation is consistent with the idea of M. A. Castello et al. 140 (a) (b) Figure 1. Neuron-like differentiated SH-SY5Y cells were treated with two oxysterols, 24-OHC and 27-OHC (10 µm) and an inducer of cholesterol dysregulation, U18666A (5 mm). (a) 27-hydroxycholesterol (27-OHC) caused a significant cell death (determined by LDH assay, arbitrary units) compared to vehicle and 24-hydroxycholesterol (24-OHC) treated cells, similar to the response induced by U18666A. (b) Filipin, a compound that binds to free intracellular cholesterol, also increases in differentiated SH-SY5Y cells treated with 27-OHC but not with 24-OHC (one-way ANOVA, p = 0.01). Together, the oxysterol 27-OHC, but not 24-OHC, appears to have a similar effect on cell survival and filipin staining as U18666A. cholesterol dysregulation contributing to early pathogenesis in late-onset AD, as we have proposed [5]. Further supporting this correlation, Pani et al. have shown that skin fibroblasts from AD patients display higher levels of filipin fluorescence [11], a result that we have confirmed in our laboratory (not shown). Overall, these results suggest that there is a continuum of filipin-sensitive cell damage that expands incrementally between healthy cognition and severe dementia. If that were the case, it might be possible to design a method to differentiate MCI sufferers according to their risk of conversion into AD by monitoring intracellular filipin levels. To be of clinical value, such a method would have to be quick and inexpensive and involve minimal risk to the patient. Thus, the use of fibroblasts is not optimal, as it involves invasive punch biopsies that are not a standard part of clinical care. In contrast, if the use of filipin could be applied to blood cells, the clinical application of our findings could be explored. To test that idea, we first determined whether filipin fluorescence can be detected in peripheral blood mononuclear cells (PBMCs) and whether it displays a range of levels that correspond to cholesterol dysregulation, as we found with SH-SY5Y cells (Figure 1). To that end, we treated them with U18666A, widely used in vitro to generate an NPC-like aberrant cholesterol phenotype [20] [21]. Figure 2 shows a dosedependent effect of U18666A on filipin staining in PBMCs, indicating that dysfunction of cholesterol metabolism can occur in PBMCs and can be measured by filipin fluorescence. Having determined that filipin-sensitive cholesterol dysregulation occurs in blood cells, we asked whether differences could exist between AD patients and cognitively healthy controls. In this case, we chose to study Blymphocytes because they are commercially available (Coriell Cell Repositories, Camden, NJ), and allow for quick confirmation of the feasibility of a filipin-based test in blood cells to determine cognitive status as well as the potential risk of MCI to AD conversion. As shown in Figure 3(a), when compared to controls, B-lymphocytes from AD patients show a significantly larger proportion of filipin-positive cells. In addition to measuring population differences between groups, we also determined the average mean fluorescence intensity in inpidual cells, as a measure of the severity of intracellular abnormalities. As shown in Figure 3(b), that average is significantly higher in AD cells, indicating that B-lymphocytes from AD patients not only show higher numbers of filipin-positive cells, but cells also display greater severity of intracellular, filipin-measurable abnormalities. 4. Discussion We have recently proposed an Adaptive Response Hypothesis of neurodegeneration to account for the etiology of late onset Alzheimer’s disease [5]. Our hypothesis states that familial and late onset forms of the disease represent etiologically different subsets of dementia and, furthermore, that dysregulation of lipids, including aberrant cholesterol oxidation patterns, is a key early pathogenic trigger of the late onset form of AD. Such dysregulation would begin at presymptomatic stages of AD, and its severity would increase with disease progression such that increasing levels of dysregulation would correlate with a worsening of cognitive status. Here, we rea- M. A. Castello et al. 141 Figure 2. Human peripheral blood mononuclear cells (PBMCs) treated with increasing concentrations of U18666A to induce lipid dysregulation also show a dose-dependent increase in the percentage of filipin-positive cells, indicating that PBMCs are also sensitive to filipin-sensitive dysregulation (one-way ANOVA, p = 0.03). (a) (b) Figure 3. (a) Flow cytometry analysis of B-lymphocytes from control and Alzheimer’s disease (AD) patients shows higher filipin fluorescence in AD cells (two-tailed t-test, p = 0.01). (b) Mean filipin fluorescence intensity of B-lymphocytes is also significantly increased in AD patients compared to control (two-tailed t-test, p = 0.02). Details on the control and AD cells used for these experiments are contained in Table 1. soned that filipin, which is already in use as a diagnostic tool in fibroblasts for other lipid dysregulation diseases, such as NPC [18], could be used to test whether such correlation exists in AD using blood cells, with the ultimate goal of designing a method of predictive value in assessing the risk of MCI to AD conversion. Our results presented here are consistent with that view, showing that the presence of intracellular abnormalities, as detected by filipin fluorescence, correlates with cellular damage (Figure 1) and is a part of AD pathology that can be readily detected in B-lymphocytes (Figure 3). The fact that cholesterol-associated cellular damage can be detected by filipin is a key novel finding. Although cholesterol-related abnormalities in peripheral cells of AD patients have been described [11] [12] [23], whether such abnormalities are reflective of pathogenesis or are instead a marker for pleiotropic effects is an important but still unanswered question with obvious implications for therapeutic intervention. In that regard, this is, to our knowledge, the first report showing that cellular damage induced by oxidized cholesterol can be measured with filipin fluorescence. Increased filipin fluorescence in response to 27-OHC cannot be accounted for by an overall increase in oxidized cholesterol levels per se, because an excess of 24-OHC, which is not cytotoxic (Figure 1(a)) does not result in an increase in the filipin signal (Figure 1(b)) [14] [15]. This suggests that filipin is detecting changes in cholesterol distribution secondary to an excess of 27-OHC, a view supported by the fact that such changes also occur and are detected by filipin in NPC [18]. The specific mechanisms by which filipin may detect cholesterol-related abnormalities beyond its established affinity for unesterified cholesterol remain M. A. Castello et al. 142 unknown and their study is outside of the scope of this report [22]. Of more direct relevance is the fact that filipin fluorescence correlates with changes elicited by U18666A and by exposure to 27-OHC, known to be upregulated in AD, both of which result in measurable cytotoxicity. We have also shown that human PBMCs are susceptible to cholesterol dysregulation as induced by U18666A and that the range of dysregulation is measurable by filipin using flow cytometry (Figure 1). These findings suggest that filipin could be of use to develop a test to compare cholesterol dysregulation levels between AD patients and cognitively functional controls. The results shown in Figure 3 provide proof of principle that this is indeed possible, showing a higher number of filipin-positive B-lymphocytes, as well as higher average mean intensity of fluorescence, in the AD population (Figure 3). Note also that cells from the control population showed greater heterogeneity compared to AD patients by both measures. Variance in the controls is predicted from a population whose cognitive function levels are heterogeneous below the threshold of cognitive impairment. In contrast, the AD population has an extremely low variance, also as expected, because their cognitive function is compromised and they have crossed the “filipin threshold”. These differences in variance are consistent with the notion that MCI patients might be separated out in terms of their risk of conversion to AD, strongly supporting a potential clinical application for filipin testing. It is also noteworthy that the choice of B-lymphocytes was determined by their commercial availability. Further work in larger clinical studies will determine differences in filipin sensitivity between these cells and different subpopulations of PBMCs, which may show comparable or superior clinical value than B-lymphocytes to diagnose cognitive status and/or identify at-risk MCI populations. In summary, we conclude that quantifying cellular filipin levels can accurately differentiate between control and AD patients in blood cells.Our findings may help define AD in the blood by focusing on markers of cellular pathology, and they provide strong preliminary data in support of larger clinical studies to test whether afilipin-based method can indeed predict the risk of AD conversion in MCI patients. Acknowledgements The authors acknowledge Dr. Kimberly Payne for help with the isolation of PBMCs and the use of flow cytometry. References [1] Alzheimer’s Association (2013) 2013 Alzheimer’s Disease Facts and Figures. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association, 9, 208-245. http://dx.doi.org/10.1016/j.jalz.2013.02.003 [2] Golde, T.E., Petrucelli, L. and Lewis, J. (2010) Targeting Aβ and Tau in Alzheimer’s Disease, an Early Interim Report. Experimental Neurology, 223, 252-266. http://dx.doi.org/10.1016/j.expneurol.2009.07.035 [3] Castellani, R.J. and Perry, G. (2012) Pathogenesis and Disease-Modifying Therapy in Alzheimer’s Disease: The Flat Line of Progress. Archives of Medical Research, 43, 694-698. http://dx.doi.org/10.1016/j.arcmed.2012.09.009 [4] Castello, M.A. and Soriano, S. (2014) On the Origin of Alzheimer’s Disease. Trials and Tribulations of the Amyloid Hypothesis. Ageing Research Reviews, 13C, 10-12. http://dx.doi.org/10.1016/j.arr.2013.10.001 [5] Castello, M.A. and Soriano, S. (2013) Rational Heterodoxy: Cholesterol Reformation of the Amyloid Doctrine. Ageing Research Reviews, 12, 282-288. http://dx.doi.org/10.1016/j.arr.2012.06.007 [6] Rodríguez-Rodríguez, E., Mateo, I., Infante, J., Llorca, J., García-Gorostiaga, I., Vázquez-Higuera, J.L. et al. (2009) Interaction between HMGCR and ABCA1 Cholesterol-Related Genes Modulates Alzheimer’s Disease Risk. Brain Research, 1280, 166-171. http://dx.doi.org/10.1016/j.brainres.2009.05.019 [7] Jones, L., Holmans, P.A., Hamshere, M.L., Harold, D., Moskvina, V., Ivanov, D., et al. (2010) Genetic Evidence Implicates the Immune System and Cholesterol Metabolism in the Aetiology of Alzheimer’s Disease. PLoS ONE, 5, Article ID: e13950. http://dx.doi.org/10.1371/journal.pone.0013950 [8] Liu, J.-P., Tang, Y., Zhou, S., Toh, B.H., McLean, C. and Li, H. (2010) Cholesterol Involvement in the Pathogenesis of Neurodegenerative Diseases. Molecular and Cellular Neuroscience, 43, 33-42. http://dx.doi.org/10.1016/j.mcn.2009.07.013 [9] Naj, A.C., Jun, G., Beecham, G.W., Wang, L.-S., Vardarajan, B.N., Buros, J., et al. (2011) Common Variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 Are Associated with Late-Onset Alzheimer’s Disease. Nature Genetics, 43, 436-441. http://dx.doi.org/10.1038/ng.801 [10] Björkhem, I. (2012) Five Decades with Oxysterols. Biochimie, 95, 448-454. M. A. Castello et al. 143 http://dx.doi.org/10.1016/j.biochi.2012.02.029 [11] Pani, A., Dessì, S., Diaz, G., La Colla, P., Abete, C., Mulas, C., et al. (2009) Altered Cholesterol Ester Cycle in Skin Fibroblasts from Patients with Alzheimer’s Disease. Journal of Alzheimer’s Disease: JAD, 18, 829-841. [12] Pani, A., Mandas, A., Diaz, G., Abete, C., Cocco, P.L., Angius, F., et al. (2009) Accumulation of Neutral Lipids in Peripheral Blood Mononuclear Cells as a Distinctive Trait of Alzheimer Patients and Asymptomatic Subjects at Risk of Disease. BMC Medicine, 7, 66. http://dx.doi.org/10.1186/1741-7015-7-66 [13] Ghribi, O., Larsen, B., Schrag, M. and Herman, M.M. (2006) High Cholesterol Content in Neurons Increases BACE, β-Amyloid, and Phosphorylated Tau Levels in Rabbit Hippocampus. Experimental Neurology, 200, 460-467. http://dx.doi.org/10.1016/j.expneurol.2006.03.019 [14] Prasanthi, J.R.P., Feist, G., Thomasson, S., Thompson, A., Schommer, E. and Ghribi, O. (2008) Differential Effects of 24-Hydroxycholesterol and 27-Hydroxycholesterol on Tyrosine Hydroxylase and α-Synuclein in Human Neuroblastoma SH-SY5Y Cells. Journal of Neurochemistry, 107, 1722-1729. http://dx.doi.org/10.1111/j.1471-4159.2008.05736.x [15] Prasanthi, J.R.P., Huls, A., Thomasson, S., Thompson, A., Schommer, E. and Ghribi, O. (2009) Differential Effects of 24-hydroxycholesterol and 27-hydroxycholesterol on β-Amyloid Precursor Protein Levels and Processing in Human Neuroblastoma SH-SY5Y Cells. Molecular Neurodegeneration, 4, 1. http://dx.doi.org/10.1186/1750-1326-4-1 [16] Burg, V.K., Grimm, H.S., Rothhaar, T.L., Grösgen, S., Hundsdörfer, B., Haupenthal, V.J., et al. (2013) Plant Sterols the Better Cholesterol in Alzheimer’s Disease? A Mechanistical Study. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 33, 16072-16087. http://dx.doi.org/10.1523/JNEUROSCI.1506-13.2013 [17] Argoff, C.E., Kaneski, C.R., Blanchette-Mackie, E.J., Comly, M., Dwyer, N.K., Brown, A., Brady, R.O. and Pentchev, P.G. (1990) Type C Niemann-Pick Disease: Documentation of Abnormal LDL Processing in Lymphocytes. Biochemical and Biophysical Research Communications, 171, 38-45. http://dx.doi.org/10.1016/0006-291X(90)91353-T [18] Distl, R., Treiber-Held, S., Albert, F., Meske, V., Harzer, K. and Ohm, T.G. (2003) Cholesterol Storage and Tau Pathology in Niemann-Pick Type C Disease in the Brain. The Journal of Pathology, 200, 104-111. http://dx.doi.org/10.1002/path.1320 [19] Tamul, K.R., Schmitz, J.L., Kane, K. and Folds, J.D. (1995) Comparison of the Effects of Ficoll-Hypaque Separation and Whole Blood Lysis on Results of Immunophenotypic Analysis of Blood and Bone Marrow Samples from Patients with Hematologic Malignancies. Clinical and Diagnostic Laboratory Immunology, 2, 337-342. [20] Koh, C.H.V. and Cheung, N.S. (2006) Cellular Mechanism of U18666A-Mediated Apoptosis in Cultured Murine Cortical Neurons: Bridging Niemann-Pick Disease Type C and Alzheimer’s Disease. Cellular Signalling, 18, 1844-1853. http://dx.doi.org/10.1016/j.cellsig.2006.04.006 [21] Cenedella, R.J. (2009) Cholesterol Synthesis Inhibitor U18666A and the Role of Sterol Metabolism and Trafficking in Numerous Pathophysiological Processes. Lipids, 44, 477-487. http://dx.doi.org/10.1007/s11745-009-3305-7 [22] Arthur, J.R., Heinecke, K.A. and Seyfried, T.N. (2011) Filipin Recognizes both GM1 and Cholesterol in GM1 Gangliosidosis Mouse brain. Journal of Lipid Research, 52, 1345-1351. http://dx.doi.org/10.1194/jlr.M012633 [23] Mandas, A., Abete, C., Putzu, P.F., la Colla, P., Dessì, S. and Pani, A. (2012) Changes in Cholesterol MetabolismRelated Gene Expression in Peripheral Blood Mononuclear Cells from Alzheimer Patients. Lipids in Health and Disease, 11, 39. http://dx.doi.org/10.1186/1476-511X-11-39
Filipin Levels as Potential Predictors of Alzheimer’s Disease Risk Michael A. Castello, Kristy D. Howard, Arthur J. Castaneda, Salvador Soriano Division of Human Anatomy, Department of Pathology and Human Anatomy, Loma Linda University School of Medicine, Loma Linda, CA, USA Email: [email protected] Received 17 July 2014; revised 20 August 2014; accepted 28 August 2014 Copyright © 2014 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract To date, therapies to prevent or treat Alzheimer’s disease (AD) have largely focused on removing excess aggregation-prone amyloid peptide Aβ from the brain, an approach that has produced disappointing clinical outcomes. An alternative hypothesis proposes that Aβ production and aggregation is a symptom of a larger, systemic disease affecting the regulation of lipids, including cholesterol. In this scenario, lipid dysregulation would likely occur early in the disease process, making it an ideal target for predicting risk of mild cognitive impairment (MCI) to AD conversion. Here, we report that levels of filipin, a fluorescent polyene macrolide widely used as a diagnostic tool for diseases of lipid dysregulation, correlate with cellular damage caused by 27-hydroxycholesterol and with dementia status in human peripheral blood cells. These results provide strong preliminary data suggesting that filipin could be of use in the development of a quick and inexpensive method to measure the risk of AD conversion in patients with MCI, supplementing existing testing strategies that focus on the consequences of Aβ accumulation. Keywords Alzheimer’s Disease, Lipid Dysregulation, Cholesterol, Mild Cognitive Impairment, Filipin Levels 1. Introduction Alzheimer’s disease (AD) is the most common form of dementia in the United States, representing around eighty percent of all cases. As the country’s sixth leading cause of death, AD currently affects 5.4 million Americans and is projected to increase to 7.7 million over the next several decades [1]. Conclusive diagnosis of AD is made post mortem, when specific pathological hallmarks are visualized in the brain: extracellular senile plaques (SPs), comprised of the amyloid-beta peptide (Aβ), and neurofibrillary tangles (NFTs), made from abnormally phos- M. A. Castello et al. 138 phorylated tau protein. To date, AD research and therapy has largely focused on removing excess aggregationprone Aβ from the brain. However, while numerous drugs have successfully reduced the burden of Aβ in the brain by removing it or preventing its aggregation, they have failed to produce significant clinical improvements [2]. These lackluster outcomes have added weight to criticism from a growing number of scientists casting doubt on the validity of the amyloid hypothesis [3] [4]. One alternative that has been proposed by ourselves and others suggests that Aβ production and aggregation is a symptom of a larger, systemic disease that affects the exquisitely-regulated balance of lipids in the brain, including cholesterol [5]. Genome-wide association studies (GWAS), as well as epidemiological and biochemical evidence, place cholesterol regulation as a likely early pathogenic trigger of late onset Alzheimer’s disease, while biochemical and histochemical studies from human autopsy brains clearly show evidence for widespread brain cholesterol dysregulation in late onset AD [6]-[9]. Of particular interest is the presence of high levels of the side-chain oxidized form of cholesterol, 27-hydroxycholesterol (27-OHC), in the AD brain. Unlike cholesterol, 27-OHC can cross the blood brain barrier, providing a mechanism to account for the link between hypercholesterolemia and the risk of AD [10]. Studies using fibroblasts and peripheral blood cells from AD patients have added weight to this argument by supplying histological and biochemical evidence of lipid dysregulation taking place in those cells [11] [12]. At the same time, APP and Aβ production has been shown to be influenced by sterols including 27-OHC [13] [16]. If dysregulation begins at presymptomatic stages of AD, and its severity increases with disease progression, it might be possible to differentiate mild cognitive impairment (MCI) sufferers according to their risk of conversion into AD by monitoring dysregulation of cholesterol or other lipids in peripheral cells. Ideally, one would want an inexpensive test that can provide quick quantification of cholesterol dysregulation using a system amenable to high throughput approaches. Here, we report that filipin, a polyene macrolide widely used as a diagnostic tool for diseases of lipid dysregulation, such as Niemann-Pick type C disease (NPC), can be of potential use to develop such a test [17] [18]. We show that filipin fluorescence, as quantified by flow cytometry, can be measured in human peripheral blood mononuclear cells (PBMCs) and that its levels correlate with dementia status. These results provide strong support for larger clinical studies to test whether our filipin-based method can in fact predict the risk of AD conversion in MCI patients, an approach that would supplement existing strategies that focus on the consequences of Aβ accumulation. 2. Materials and Methods 2.1. Reagents U18666A (Sigma-Aldrich, St. Louis, MO) was stored in aliquots at -80˚C as an aqueous solution at a concentration of 4.7163 M (2 mg/ml). Working concentrations were prepared in culture medium immediately prior to experiments. 24-hydroxycholesterol (24-OHC) and 27-hydroxycholesterol (27-OHC) (Cayman Chemical, Ann Arbor, MI) were prepared as 10 mm stock solutions in 100% ethanol (Sigma-Aldrich, St. Louis, MO) and stored in aliquots at −80˚C. Working concentrations were prepared in culture medium immediately prior to experiments. Except where otherwise noted, cells were treated with media containing 5 mm U18666A, 10 µm 24-OHC, or 10 µm 27-OHC and incubated for 16 hours at 37˚C. 2.2. Cell Culture and Fluorescence Microscopy B-lymphocytes were obtained from Coriell cell repository (Camden, NJ; Table 1) and cultured in RPMI (Life Technologies, Carlsbad, CA) with 15% FBS (Atalanta Biologicals, Flowery Branch, GA) and 1% pen/strep (Life Technologies, Carlsbad, CA). According to information available from Coriell, AD patients were not known to be homozygous for ApoE4. SH-SY5Y human neuroblastoma cells (CRL-2266, American Type Culture Collection) were cultured in DMEM (Life Technologies, Carlsbad, CA) with 15% FBS and 1% pen/strep. For neuronal differentiation, cells were plated in 6-well plates at 400,000 cells/well (day 1). After 24 hrs (day 2), media was changed to DMEM with 3.75% FBS, 1% pen/strep, and 10 µm retinoic acid (RA; Sigma-Aldrich, St. Louis, MO). On day 5, media was discarded and replaced with fresh DMEM, again with 3.75% FBS, 1% pen/strep, and 10 µm RA. On day 6, M. A. Castello et al. 139 Table 1. Cells acquired from Coriell cell repository. According to the information available from Coriell, AD patients were not known to be homozygous for ApoE4. Cells Acquired from Coriell Cell Repository Catalog ID Group Age Cell Type AG08526 AD 61 B-Lymphocyte AG21157 AD 61 B-Lymphocyte AG06204 AD 67 B-Lymphocyte AG21159 AD 69 B-Lymphocyte AG08242 AD 72 B-Lymphocyte AG10986 Control 82 B-Lymphocyte AG10955 Control 89 B-Lymphocyte AG08897 Control 61 B-Lymphocyte media was changed to DMEM without FBS, and containing 10 µm RA with 50 ng/ml of brain-derived neurotrophic factor (BDNF; Sigma-Aldrich, St. Louis, MO). Differentiated cells were experimentally treated on day 8. Peripheral blood mononuclear cells (PBMCs) from adult donors were obtained from Leuko-pak leukocyte filters (Fenwal Laboratories, Lake Zurich, IL) and donated by the Blood Processing and Quality Control Lifestream (San Bernardino, CA). Ficoll-Hypaque (GE Healthcare, Pittsburgh, PA) and red blood cell lysis density gradient centrifugation were used for PBMC isolation exactly as described [19]. 2.3. Flow Cytometry For FACS analysis, cells were fixed in paraformaldehyde (PFA) at room temperature (RT; peripheral blood cells, 1% PFA 30 min at RT; B-lymphocytes, 4% PFA 1 hr at RT) and treated with filipin for 30 min at RT (peripheral blood cells, 50 μg/ml filipin; B-lymphocytes, 100 μg/ml filipin). Flow cytometry was performed using a MACSQuant Analyzer (Miltenyi Biotec, Bergisch Gladbach, Germany) with excitation at 350 nm. Mean fluorescence intensity was calculated with FlowJo software (TreeStar, Inc., Ashland, OR). Average percentage of filipin-positive to total number of cells was determined in FlowJo using flipin fluorescence in cells from control patients (B-lymphocytes) or vehicle-treated cells (SH-SY5Y and PBMCs) to determine cut-offs (as described in Figure S1). Statistical analysis of the percentages was performed in SPSS Statistics (version 22, IBM Corp.) using one-way ANOVA and Bonferroni post-hoc analysis. 3. Results To assess the impact of cholesterol dysregulation on cytotoxicity, we treated neuron-differentiated SH-SY5Y cells with side-chain oxidized cholesterol forms 24-OHC or 27-OHC, and measured extracellular lactate dehydrogenase (LDH) as an indicator of cell damage. In addition, we treated cells with U18666A, a compound that inhibits cholesterol synthesis and intracellular trafficking and leads to apoptosis in a variety of cell lines, including neurons, thus serving as positive control for cholesterol-associated cytotoxicity [20] [21]. As shown in Figure 1(a), treatment of cells with 27-OHC, but not 24-OHC, led to significantly higher levels of LDH release than in untreated control cells, in agreement with previous work from the Ghribi laboratory [15]. Note also that the cytotoxic impact of 27-OHC was comparable to that seen in cells treated with U18666. Next, we assessed the correlation between the cellular damage, as detected by LDH release, and levels of filipin fluorescence. Neuron-differentiated SH-SY5Y cells were treated as above with 24-OHC, 27-OHC, or U18666A, and fluorescence from filipin was measured by fluorescence-activated cell sorting (FACS). As seen in Figure 1(b), 27-OHC, but not 24-OHC, led to a statistically significant increase in filipin staining that was comparable to the impact of U18666A. This differential result indicates that, in our SH-SY5Y neuronal model, filipin fluorescence does not correlate with overall levels of oxidized cholesterol, because 24-OHC does not lead to significant changes in filipin signal. Rather, filipin fluorescence correlates with the cytotoxicity status that accompanies the changes in subcellular distribution of the lipids that it detects [22]. This correlation is consistent with the idea of M. A. Castello et al. 140 (a) (b) Figure 1. Neuron-like differentiated SH-SY5Y cells were treated with two oxysterols, 24-OHC and 27-OHC (10 µm) and an inducer of cholesterol dysregulation, U18666A (5 mm). (a) 27-hydroxycholesterol (27-OHC) caused a significant cell death (determined by LDH assay, arbitrary units) compared to vehicle and 24-hydroxycholesterol (24-OHC) treated cells, similar to the response induced by U18666A. (b) Filipin, a compound that binds to free intracellular cholesterol, also increases in differentiated SH-SY5Y cells treated with 27-OHC but not with 24-OHC (one-way ANOVA, p = 0.01). Together, the oxysterol 27-OHC, but not 24-OHC, appears to have a similar effect on cell survival and filipin staining as U18666A. cholesterol dysregulation contributing to early pathogenesis in late-onset AD, as we have proposed [5]. Further supporting this correlation, Pani et al. have shown that skin fibroblasts from AD patients display higher levels of filipin fluorescence [11], a result that we have confirmed in our laboratory (not shown). Overall, these results suggest that there is a continuum of filipin-sensitive cell damage that expands incrementally between healthy cognition and severe dementia. If that were the case, it might be possible to design a method to differentiate MCI sufferers according to their risk of conversion into AD by monitoring intracellular filipin levels. To be of clinical value, such a method would have to be quick and inexpensive and involve minimal risk to the patient. Thus, the use of fibroblasts is not optimal, as it involves invasive punch biopsies that are not a standard part of clinical care. In contrast, if the use of filipin could be applied to blood cells, the clinical application of our findings could be explored. To test that idea, we first determined whether filipin fluorescence can be detected in peripheral blood mononuclear cells (PBMCs) and whether it displays a range of levels that correspond to cholesterol dysregulation, as we found with SH-SY5Y cells (Figure 1). To that end, we treated them with U18666A, widely used in vitro to generate an NPC-like aberrant cholesterol phenotype [20] [21]. Figure 2 shows a dosedependent effect of U18666A on filipin staining in PBMCs, indicating that dysfunction of cholesterol metabolism can occur in PBMCs and can be measured by filipin fluorescence. Having determined that filipin-sensitive cholesterol dysregulation occurs in blood cells, we asked whether differences could exist between AD patients and cognitively healthy controls. In this case, we chose to study Blymphocytes because they are commercially available (Coriell Cell Repositories, Camden, NJ), and allow for quick confirmation of the feasibility of a filipin-based test in blood cells to determine cognitive status as well as the potential risk of MCI to AD conversion. As shown in Figure 3(a), when compared to controls, B-lymphocytes from AD patients show a significantly larger proportion of filipin-positive cells. In addition to measuring population differences between groups, we also determined the average mean fluorescence intensity in inpidual cells, as a measure of the severity of intracellular abnormalities. As shown in Figure 3(b), that average is significantly higher in AD cells, indicating that B-lymphocytes from AD patients not only show higher numbers of filipin-positive cells, but cells also display greater severity of intracellular, filipin-measurable abnormalities. 4. Discussion We have recently proposed an Adaptive Response Hypothesis of neurodegeneration to account for the etiology of late onset Alzheimer’s disease [5]. Our hypothesis states that familial and late onset forms of the disease represent etiologically different subsets of dementia and, furthermore, that dysregulation of lipids, including aberrant cholesterol oxidation patterns, is a key early pathogenic trigger of the late onset form of AD. Such dysregulation would begin at presymptomatic stages of AD, and its severity would increase with disease progression such that increasing levels of dysregulation would correlate with a worsening of cognitive status. Here, we rea- M. A. Castello et al. 141 Figure 2. Human peripheral blood mononuclear cells (PBMCs) treated with increasing concentrations of U18666A to induce lipid dysregulation also show a dose-dependent increase in the percentage of filipin-positive cells, indicating that PBMCs are also sensitive to filipin-sensitive dysregulation (one-way ANOVA, p = 0.03). (a) (b) Figure 3. (a) Flow cytometry analysis of B-lymphocytes from control and Alzheimer’s disease (AD) patients shows higher filipin fluorescence in AD cells (two-tailed t-test, p = 0.01). (b) Mean filipin fluorescence intensity of B-lymphocytes is also significantly increased in AD patients compared to control (two-tailed t-test, p = 0.02). Details on the control and AD cells used for these experiments are contained in Table 1. soned that filipin, which is already in use as a diagnostic tool in fibroblasts for other lipid dysregulation diseases, such as NPC [18], could be used to test whether such correlation exists in AD using blood cells, with the ultimate goal of designing a method of predictive value in assessing the risk of MCI to AD conversion. Our results presented here are consistent with that view, showing that the presence of intracellular abnormalities, as detected by filipin fluorescence, correlates with cellular damage (Figure 1) and is a part of AD pathology that can be readily detected in B-lymphocytes (Figure 3). The fact that cholesterol-associated cellular damage can be detected by filipin is a key novel finding. Although cholesterol-related abnormalities in peripheral cells of AD patients have been described [11] [12] [23], whether such abnormalities are reflective of pathogenesis or are instead a marker for pleiotropic effects is an important but still unanswered question with obvious implications for therapeutic intervention. In that regard, this is, to our knowledge, the first report showing that cellular damage induced by oxidized cholesterol can be measured with filipin fluorescence. Increased filipin fluorescence in response to 27-OHC cannot be accounted for by an overall increase in oxidized cholesterol levels per se, because an excess of 24-OHC, which is not cytotoxic (Figure 1(a)) does not result in an increase in the filipin signal (Figure 1(b)) [14] [15]. This suggests that filipin is detecting changes in cholesterol distribution secondary to an excess of 27-OHC, a view supported by the fact that such changes also occur and are detected by filipin in NPC [18]. The specific mechanisms by which filipin may detect cholesterol-related abnormalities beyond its established affinity for unesterified cholesterol remain M. A. Castello et al. 142 unknown and their study is outside of the scope of this report [22]. Of more direct relevance is the fact that filipin fluorescence correlates with changes elicited by U18666A and by exposure to 27-OHC, known to be upregulated in AD, both of which result in measurable cytotoxicity. We have also shown that human PBMCs are susceptible to cholesterol dysregulation as induced by U18666A and that the range of dysregulation is measurable by filipin using flow cytometry (Figure 1). These findings suggest that filipin could be of use to develop a test to compare cholesterol dysregulation levels between AD patients and cognitively functional controls. The results shown in Figure 3 provide proof of principle that this is indeed possible, showing a higher number of filipin-positive B-lymphocytes, as well as higher average mean intensity of fluorescence, in the AD population (Figure 3). Note also that cells from the control population showed greater heterogeneity compared to AD patients by both measures. Variance in the controls is predicted from a population whose cognitive function levels are heterogeneous below the threshold of cognitive impairment. In contrast, the AD population has an extremely low variance, also as expected, because their cognitive function is compromised and they have crossed the “filipin threshold”. These differences in variance are consistent with the notion that MCI patients might be separated out in terms of their risk of conversion to AD, strongly supporting a potential clinical application for filipin testing. It is also noteworthy that the choice of B-lymphocytes was determined by their commercial availability. Further work in larger clinical studies will determine differences in filipin sensitivity between these cells and different subpopulations of PBMCs, which may show comparable or superior clinical value than B-lymphocytes to diagnose cognitive status and/or identify at-risk MCI populations. In summary, we conclude that quantifying cellular filipin levels can accurately differentiate between control and AD patients in blood cells.Our findings may help define AD in the blood by focusing on markers of cellular pathology, and they provide strong preliminary data in support of larger clinical studies to test whether afilipin-based method can indeed predict the risk of AD conversion in MCI patients. Acknowledgements The authors acknowledge Dr. Kimberly Payne for help with the isolation of PBMCs and the use of flow cytometry. References [1] Alzheimer’s Association (2013) 2013 Alzheimer’s Disease Facts and Figures. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association, 9, 208-245. http://dx.doi.org/10.1016/j.jalz.2013.02.003 [2] Golde, T.E., Petrucelli, L. and Lewis, J. (2010) Targeting Aβ and Tau in Alzheimer’s Disease, an Early Interim Report. Experimental Neurology, 223, 252-266. http://dx.doi.org/10.1016/j.expneurol.2009.07.035 [3] Castellani, R.J. and Perry, G. (2012) Pathogenesis and Disease-Modifying Therapy in Alzheimer’s Disease: The Flat Line of Progress. Archives of Medical Research, 43, 694-698. http://dx.doi.org/10.1016/j.arcmed.2012.09.009 [4] Castello, M.A. and Soriano, S. (2014) On the Origin of Alzheimer’s Disease. Trials and Tribulations of the Amyloid Hypothesis. Ageing Research Reviews, 13C, 10-12. http://dx.doi.org/10.1016/j.arr.2013.10.001 [5] Castello, M.A. and Soriano, S. (2013) Rational Heterodoxy: Cholesterol Reformation of the Amyloid Doctrine. Ageing Research Reviews, 12, 282-288. http://dx.doi.org/10.1016/j.arr.2012.06.007 [6] Rodríguez-Rodríguez, E., Mateo, I., Infante, J., Llorca, J., García-Gorostiaga, I., Vázquez-Higuera, J.L. et al. (2009) Interaction between HMGCR and ABCA1 Cholesterol-Related Genes Modulates Alzheimer’s Disease Risk. Brain Research, 1280, 166-171. http://dx.doi.org/10.1016/j.brainres.2009.05.019 [7] Jones, L., Holmans, P.A., Hamshere, M.L., Harold, D., Moskvina, V., Ivanov, D., et al. (2010) Genetic Evidence Implicates the Immune System and Cholesterol Metabolism in the Aetiology of Alzheimer’s Disease. PLoS ONE, 5, Article ID: e13950. http://dx.doi.org/10.1371/journal.pone.0013950 [8] Liu, J.-P., Tang, Y., Zhou, S., Toh, B.H., McLean, C. and Li, H. (2010) Cholesterol Involvement in the Pathogenesis of Neurodegenerative Diseases. Molecular and Cellular Neuroscience, 43, 33-42. http://dx.doi.org/10.1016/j.mcn.2009.07.013 [9] Naj, A.C., Jun, G., Beecham, G.W., Wang, L.-S., Vardarajan, B.N., Buros, J., et al. (2011) Common Variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 Are Associated with Late-Onset Alzheimer’s Disease. Nature Genetics, 43, 436-441. http://dx.doi.org/10.1038/ng.801 [10] Björkhem, I. (2012) Five Decades with Oxysterols. Biochimie, 95, 448-454. M. A. Castello et al. 143 http://dx.doi.org/10.1016/j.biochi.2012.02.029 [11] Pani, A., Dessì, S., Diaz, G., La Colla, P., Abete, C., Mulas, C., et al. (2009) Altered Cholesterol Ester Cycle in Skin Fibroblasts from Patients with Alzheimer’s Disease. Journal of Alzheimer’s Disease: JAD, 18, 829-841. [12] Pani, A., Mandas, A., Diaz, G., Abete, C., Cocco, P.L., Angius, F., et al. (2009) Accumulation of Neutral Lipids in Peripheral Blood Mononuclear Cells as a Distinctive Trait of Alzheimer Patients and Asymptomatic Subjects at Risk of Disease. BMC Medicine, 7, 66. http://dx.doi.org/10.1186/1741-7015-7-66 [13] Ghribi, O., Larsen, B., Schrag, M. and Herman, M.M. (2006) High Cholesterol Content in Neurons Increases BACE, β-Amyloid, and Phosphorylated Tau Levels in Rabbit Hippocampus. Experimental Neurology, 200, 460-467. http://dx.doi.org/10.1016/j.expneurol.2006.03.019 [14] Prasanthi, J.R.P., Feist, G., Thomasson, S., Thompson, A., Schommer, E. and Ghribi, O. (2008) Differential Effects of 24-Hydroxycholesterol and 27-Hydroxycholesterol on Tyrosine Hydroxylase and α-Synuclein in Human Neuroblastoma SH-SY5Y Cells. Journal of Neurochemistry, 107, 1722-1729. http://dx.doi.org/10.1111/j.1471-4159.2008.05736.x [15] Prasanthi, J.R.P., Huls, A., Thomasson, S., Thompson, A., Schommer, E. and Ghribi, O. (2009) Differential Effects of 24-hydroxycholesterol and 27-hydroxycholesterol on β-Amyloid Precursor Protein Levels and Processing in Human Neuroblastoma SH-SY5Y Cells. Molecular Neurodegeneration, 4, 1. http://dx.doi.org/10.1186/1750-1326-4-1 [16] Burg, V.K., Grimm, H.S., Rothhaar, T.L., Grösgen, S., Hundsdörfer, B., Haupenthal, V.J., et al. (2013) Plant Sterols the Better Cholesterol in Alzheimer’s Disease? A Mechanistical Study. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 33, 16072-16087. http://dx.doi.org/10.1523/JNEUROSCI.1506-13.2013 [17] Argoff, C.E., Kaneski, C.R., Blanchette-Mackie, E.J., Comly, M., Dwyer, N.K., Brown, A., Brady, R.O. and Pentchev, P.G. (1990) Type C Niemann-Pick Disease: Documentation of Abnormal LDL Processing in Lymphocytes. Biochemical and Biophysical Research Communications, 171, 38-45. http://dx.doi.org/10.1016/0006-291X(90)91353-T [18] Distl, R., Treiber-Held, S., Albert, F., Meske, V., Harzer, K. and Ohm, T.G. (2003) Cholesterol Storage and Tau Pathology in Niemann-Pick Type C Disease in the Brain. The Journal of Pathology, 200, 104-111. http://dx.doi.org/10.1002/path.1320 [19] Tamul, K.R., Schmitz, J.L., Kane, K. and Folds, J.D. (1995) Comparison of the Effects of Ficoll-Hypaque Separation and Whole Blood Lysis on Results of Immunophenotypic Analysis of Blood and Bone Marrow Samples from Patients with Hematologic Malignancies. Clinical and Diagnostic Laboratory Immunology, 2, 337-342. [20] Koh, C.H.V. and Cheung, N.S. (2006) Cellular Mechanism of U18666A-Mediated Apoptosis in Cultured Murine Cortical Neurons: Bridging Niemann-Pick Disease Type C and Alzheimer’s Disease. Cellular Signalling, 18, 1844-1853. http://dx.doi.org/10.1016/j.cellsig.2006.04.006 [21] Cenedella, R.J. (2009) Cholesterol Synthesis Inhibitor U18666A and the Role of Sterol Metabolism and Trafficking in Numerous Pathophysiological Processes. Lipids, 44, 477-487. http://dx.doi.org/10.1007/s11745-009-3305-7 [22] Arthur, J.R., Heinecke, K.A. and Seyfried, T.N. (2011) Filipin Recognizes both GM1 and Cholesterol in GM1 Gangliosidosis Mouse brain. Journal of Lipid Research, 52, 1345-1351. http://dx.doi.org/10.1194/jlr.M012633 [23] Mandas, A., Abete, C., Putzu, P.F., la Colla, P., Dessì, S. and Pani, A. (2012) Changes in Cholesterol MetabolismRelated Gene Expression in Peripheral Blood Mononuclear Cells from Alzheimer Patients. Lipids in Health and Disease, 11, 39. http://dx.doi.org/10.1186/1476-511X-11-39[Filipin Levels as Potential Predictors ofAlzheimer’s Disease Risk] as of
Filipin Levels as Potential Predictors of Alzheimer’s Disease Risk Michael A. Castello, Kristy D. Howard, Arthur J. Castaneda, Salvador Soriano Division of Human Anatomy, Department of Pathology and Human Anatomy, Loma Linda University School of Medicine, Loma Linda, CA, USA Email: [email protected] Received 17 July 2014; revised 20 August 2014; accepted 28 August 2014 Copyright © 2014 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract To date, therapies to prevent or treat Alzheimer’s disease (AD) have largely focused on removing excess aggregation-prone amyloid peptide Aβ from the brain, an approach that has produced disappointing clinical outcomes. An alternative hypothesis proposes that Aβ production and aggregation is a symptom of a larger, systemic disease affecting the regulation of lipids, including cholesterol. In this scenario, lipid dysregulation would likely occur early in the disease process, making it an ideal target for predicting risk of mild cognitive impairment (MCI) to AD conversion. Here, we report that levels of filipin, a fluorescent polyene macrolide widely used as a diagnostic tool for diseases of lipid dysregulation, correlate with cellular damage caused by 27-hydroxycholesterol and with dementia status in human peripheral blood cells. These results provide strong preliminary data suggesting that filipin could be of use in the development of a quick and inexpensive method to measure the risk of AD conversion in patients with MCI, supplementing existing testing strategies that focus on the consequences of Aβ accumulation. Keywords Alzheimer’s Disease, Lipid Dysregulation, Cholesterol, Mild Cognitive Impairment, Filipin Levels 1. Introduction Alzheimer’s disease (AD) is the most common form of dementia in the United States, representing around eighty percent of all cases. As the country’s sixth leading cause of death, AD currently affects 5.4 million Americans and is projected to increase to 7.7 million over the next several decades [1]. Conclusive diagnosis of AD is made post mortem, when specific pathological hallmarks are visualized in the brain: extracellular senile plaques (SPs), comprised of the amyloid-beta peptide (Aβ), and neurofibrillary tangles (NFTs), made from abnormally phos- M. A. Castello et al. 138 phorylated tau protein. To date, AD research and therapy has largely focused on removing excess aggregationprone Aβ from the brain. However, while numerous drugs have successfully reduced the burden of Aβ in the brain by removing it or preventing its aggregation, they have failed to produce significant clinical improvements [2]. These lackluster outcomes have added weight to criticism from a growing number of scientists casting doubt on the validity of the amyloid hypothesis [3] [4]. One alternative that has been proposed by ourselves and others suggests that Aβ production and aggregation is a symptom of a larger, systemic disease that affects the exquisitely-regulated balance of lipids in the brain, including cholesterol [5]. Genome-wide association studies (GWAS), as well as epidemiological and biochemical evidence, place cholesterol regulation as a likely early pathogenic trigger of late onset Alzheimer’s disease, while biochemical and histochemical studies from human autopsy brains clearly show evidence for widespread brain cholesterol dysregulation in late onset AD [6]-[9]. Of particular interest is the presence of high levels of the side-chain oxidized form of cholesterol, 27-hydroxycholesterol (27-OHC), in the AD brain. Unlike cholesterol, 27-OHC can cross the blood brain barrier, providing a mechanism to account for the link between hypercholesterolemia and the risk of AD [10]. Studies using fibroblasts and peripheral blood cells from AD patients have added weight to this argument by supplying histological and biochemical evidence of lipid dysregulation taking place in those cells [11] [12]. At the same time, APP and Aβ production has been shown to be influenced by sterols including 27-OHC [13] [16]. If dysregulation begins at presymptomatic stages of AD, and its severity increases with disease progression, it might be possible to differentiate mild cognitive impairment (MCI) sufferers according to their risk of conversion into AD by monitoring dysregulation of cholesterol or other lipids in peripheral cells. Ideally, one would want an inexpensive test that can provide quick quantification of cholesterol dysregulation using a system amenable to high throughput approaches. Here, we report that filipin, a polyene macrolide widely used as a diagnostic tool for diseases of lipid dysregulation, such as Niemann-Pick type C disease (NPC), can be of potential use to develop such a test [17] [18]. We show that filipin fluorescence, as quantified by flow cytometry, can be measured in human peripheral blood mononuclear cells (PBMCs) and that its levels correlate with dementia status. These results provide strong support for larger clinical studies to test whether our filipin-based method can in fact predict the risk of AD conversion in MCI patients, an approach that would supplement existing strategies that focus on the consequences of Aβ accumulation. 2. Materials and Methods 2.1. Reagents U18666A (Sigma-Aldrich, St. Louis, MO) was stored in aliquots at -80˚C as an aqueous solution at a concentration of 4.7163 M (2 mg/ml). Working concentrations were prepared in culture medium immediately prior to experiments. 24-hydroxycholesterol (24-OHC) and 27-hydroxycholesterol (27-OHC) (Cayman Chemical, Ann Arbor, MI) were prepared as 10 mm stock solutions in 100% ethanol (Sigma-Aldrich, St. Louis, MO) and stored in aliquots at −80˚C. Working concentrations were prepared in culture medium immediately prior to experiments. Except where otherwise noted, cells were treated with media containing 5 mm U18666A, 10 µm 24-OHC, or 10 µm 27-OHC and incubated for 16 hours at 37˚C. 2.2. Cell Culture and Fluorescence Microscopy B-lymphocytes were obtained from Coriell cell repository (Camden, NJ; Table 1) and cultured in RPMI (Life Technologies, Carlsbad, CA) with 15% FBS (Atalanta Biologicals, Flowery Branch, GA) and 1% pen/strep (Life Technologies, Carlsbad, CA). According to information available from Coriell, AD patients were not known to be homozygous for ApoE4. SH-SY5Y human neuroblastoma cells (CRL-2266, American Type Culture Collection) were cultured in DMEM (Life Technologies, Carlsbad, CA) with 15% FBS and 1% pen/strep. For neuronal differentiation, cells were plated in 6-well plates at 400,000 cells/well (day 1). After 24 hrs (day 2), media was changed to DMEM with 3.75% FBS, 1% pen/strep, and 10 µm retinoic acid (RA; Sigma-Aldrich, St. Louis, MO). On day 5, media was discarded and replaced with fresh DMEM, again with 3.75% FBS, 1% pen/strep, and 10 µm RA. On day 6, M. A. Castello et al. 139 Table 1. Cells acquired from Coriell cell repository. According to the information available from Coriell, AD patients were not known to be homozygous for ApoE4. Cells Acquired from Coriell Cell Repository Catalog ID Group Age Cell Type AG08526 AD 61 B-Lymphocyte AG21157 AD 61 B-Lymphocyte AG06204 AD 67 B-Lymphocyte AG21159 AD 69 B-Lymphocyte AG08242 AD 72 B-Lymphocyte AG10986 Control 82 B-Lymphocyte AG10955 Control 89 B-Lymphocyte AG08897 Control 61 B-Lymphocyte media was changed to DMEM without FBS, and containing 10 µm RA with 50 ng/ml of brain-derived neurotrophic factor (BDNF; Sigma-Aldrich, St. Louis, MO). Differentiated cells were experimentally treated on day 8. Peripheral blood mononuclear cells (PBMCs) from adult donors were obtained from Leuko-pak leukocyte filters (Fenwal Laboratories, Lake Zurich, IL) and donated by the Blood Processing and Quality Control Lifestream (San Bernardino, CA). Ficoll-Hypaque (GE Healthcare, Pittsburgh, PA) and red blood cell lysis density gradient centrifugation were used for PBMC isolation exactly as described [19]. 2.3. Flow Cytometry For FACS analysis, cells were fixed in paraformaldehyde (PFA) at room temperature (RT; peripheral blood cells, 1% PFA 30 min at RT; B-lymphocytes, 4% PFA 1 hr at RT) and treated with filipin for 30 min at RT (peripheral blood cells, 50 μg/ml filipin; B-lymphocytes, 100 μg/ml filipin). Flow cytometry was performed using a MACSQuant Analyzer (Miltenyi Biotec, Bergisch Gladbach, Germany) with excitation at 350 nm. Mean fluorescence intensity was calculated with FlowJo software (TreeStar, Inc., Ashland, OR). Average percentage of filipin-positive to total number of cells was determined in FlowJo using flipin fluorescence in cells from control patients (B-lymphocytes) or vehicle-treated cells (SH-SY5Y and PBMCs) to determine cut-offs (as described in Figure S1). Statistical analysis of the percentages was performed in SPSS Statistics (version 22, IBM Corp.) using one-way ANOVA and Bonferroni post-hoc analysis. 3. Results To assess the impact of cholesterol dysregulation on cytotoxicity, we treated neuron-differentiated SH-SY5Y cells with side-chain oxidized cholesterol forms 24-OHC or 27-OHC, and measured extracellular lactate dehydrogenase (LDH) as an indicator of cell damage. In addition, we treated cells with U18666A, a compound that inhibits cholesterol synthesis and intracellular trafficking and leads to apoptosis in a variety of cell lines, including neurons, thus serving as positive control for cholesterol-associated cytotoxicity [20] [21]. As shown in Figure 1(a), treatment of cells with 27-OHC, but not 24-OHC, led to significantly higher levels of LDH release than in untreated control cells, in agreement with previous work from the Ghribi laboratory [15]. Note also that the cytotoxic impact of 27-OHC was comparable to that seen in cells treated with U18666. Next, we assessed the correlation between the cellular damage, as detected by LDH release, and levels of filipin fluorescence. Neuron-differentiated SH-SY5Y cells were treated as above with 24-OHC, 27-OHC, or U18666A, and fluorescence from filipin was measured by fluorescence-activated cell sorting (FACS). As seen in Figure 1(b), 27-OHC, but not 24-OHC, led to a statistically significant increase in filipin staining that was comparable to the impact of U18666A. This differential result indicates that, in our SH-SY5Y neuronal model, filipin fluorescence does not correlate with overall levels of oxidized cholesterol, because 24-OHC does not lead to significant changes in filipin signal. Rather, filipin fluorescence correlates with the cytotoxicity status that accompanies the changes in subcellular distribution of the lipids that it detects [22]. This correlation is consistent with the idea of M. A. Castello et al. 140 (a) (b) Figure 1. Neuron-like differentiated SH-SY5Y cells were treated with two oxysterols, 24-OHC and 27-OHC (10 µm) and an inducer of cholesterol dysregulation, U18666A (5 mm). (a) 27-hydroxycholesterol (27-OHC) caused a significant cell death (determined by LDH assay, arbitrary units) compared to vehicle and 24-hydroxycholesterol (24-OHC) treated cells, similar to the response induced by U18666A. (b) Filipin, a compound that binds to free intracellular cholesterol, also increases in differentiated SH-SY5Y cells treated with 27-OHC but not with 24-OHC (one-way ANOVA, p = 0.01). Together, the oxysterol 27-OHC, but not 24-OHC, appears to have a similar effect on cell survival and filipin staining as U18666A. cholesterol dysregulation contributing to early pathogenesis in late-onset AD, as we have proposed [5]. Further supporting this correlation, Pani et al. have shown that skin fibroblasts from AD patients display higher levels of filipin fluorescence [11], a result that we have confirmed in our laboratory (not shown). Overall, these results suggest that there is a continuum of filipin-sensitive cell damage that expands incrementally between healthy cognition and severe dementia. If that were the case, it might be possible to design a method to differentiate MCI sufferers according to their risk of conversion into AD by monitoring intracellular filipin levels. To be of clinical value, such a method would have to be quick and inexpensive and involve minimal risk to the patient. Thus, the use of fibroblasts is not optimal, as it involves invasive punch biopsies that are not a standard part of clinical care. In contrast, if the use of filipin could be applied to blood cells, the clinical application of our findings could be explored. To test that idea, we first determined whether filipin fluorescence can be detected in peripheral blood mononuclear cells (PBMCs) and whether it displays a range of levels that correspond to cholesterol dysregulation, as we found with SH-SY5Y cells (Figure 1). To that end, we treated them with U18666A, widely used in vitro to generate an NPC-like aberrant cholesterol phenotype [20] [21]. Figure 2 shows a dosedependent effect of U18666A on filipin staining in PBMCs, indicating that dysfunction of cholesterol metabolism can occur in PBMCs and can be measured by filipin fluorescence. Having determined that filipin-sensitive cholesterol dysregulation occurs in blood cells, we asked whether differences could exist between AD patients and cognitively healthy controls. In this case, we chose to study Blymphocytes because they are commercially available (Coriell Cell Repositories, Camden, NJ), and allow for quick confirmation of the feasibility of a filipin-based test in blood cells to determine cognitive status as well as the potential risk of MCI to AD conversion. As shown in Figure 3(a), when compared to controls, B-lymphocytes from AD patients show a significantly larger proportion of filipin-positive cells. In addition to measuring population differences between groups, we also determined the average mean fluorescence intensity in inpidual cells, as a measure of the severity of intracellular abnormalities. As shown in Figure 3(b), that average is significantly higher in AD cells, indicating that B-lymphocytes from AD patients not only show higher numbers of filipin-positive cells, but cells also display greater severity of intracellular, filipin-measurable abnormalities. 4. Discussion We have recently proposed an Adaptive Response Hypothesis of neurodegeneration to account for the etiology of late onset Alzheimer’s disease [5]. Our hypothesis states that familial and late onset forms of the disease represent etiologically different subsets of dementia and, furthermore, that dysregulation of lipids, including aberrant cholesterol oxidation patterns, is a key early pathogenic trigger of the late onset form of AD. Such dysregulation would begin at presymptomatic stages of AD, and its severity would increase with disease progression such that increasing levels of dysregulation would correlate with a worsening of cognitive status. Here, we rea- M. A. Castello et al. 141 Figure 2. Human peripheral blood mononuclear cells (PBMCs) treated with increasing concentrations of U18666A to induce lipid dysregulation also show a dose-dependent increase in the percentage of filipin-positive cells, indicating that PBMCs are also sensitive to filipin-sensitive dysregulation (one-way ANOVA, p = 0.03). (a) (b) Figure 3. (a) Flow cytometry analysis of B-lymphocytes from control and Alzheimer’s disease (AD) patients shows higher filipin fluorescence in AD cells (two-tailed t-test, p = 0.01). (b) Mean filipin fluorescence intensity of B-lymphocytes is also significantly increased in AD patients compared to control (two-tailed t-test, p = 0.02). Details on the control and AD cells used for these experiments are contained in Table 1. soned that filipin, which is already in use as a diagnostic tool in fibroblasts for other lipid dysregulation diseases, such as NPC [18], could be used to test whether such correlation exists in AD using blood cells, with the ultimate goal of designing a method of predictive value in assessing the risk of MCI to AD conversion. Our results presented here are consistent with that view, showing that the presence of intracellular abnormalities, as detected by filipin fluorescence, correlates with cellular damage (Figure 1) and is a part of AD pathology that can be readily detected in B-lymphocytes (Figure 3). The fact that cholesterol-associated cellular damage can be detected by filipin is a key novel finding. Although cholesterol-related abnormalities in peripheral cells of AD patients have been described [11] [12] [23], whether such abnormalities are reflective of pathogenesis or are instead a marker for pleiotropic effects is an important but still unanswered question with obvious implications for therapeutic intervention. In that regard, this is, to our knowledge, the first report showing that cellular damage induced by oxidized cholesterol can be measured with filipin fluorescence. Increased filipin fluorescence in response to 27-OHC cannot be accounted for by an overall increase in oxidized cholesterol levels per se, because an excess of 24-OHC, which is not cytotoxic (Figure 1(a)) does not result in an increase in the filipin signal (Figure 1(b)) [14] [15]. This suggests that filipin is detecting changes in cholesterol distribution secondary to an excess of 27-OHC, a view supported by the fact that such changes also occur and are detected by filipin in NPC [18]. The specific mechanisms by which filipin may detect cholesterol-related abnormalities beyond its established affinity for unesterified cholesterol remain M. A. Castello et al. 142 unknown and their study is outside of the scope of this report [22]. Of more direct relevance is the fact that filipin fluorescence correlates with changes elicited by U18666A and by exposure to 27-OHC, known to be upregulated in AD, both of which result in measurable cytotoxicity. We have also shown that human PBMCs are susceptible to cholesterol dysregulation as induced by U18666A and that the range of dysregulation is measurable by filipin using flow cytometry (Figure 1). These findings suggest that filipin could be of use to develop a test to compare cholesterol dysregulation levels between AD patients and cognitively functional controls. The results shown in Figure 3 provide proof of principle that this is indeed possible, showing a higher number of filipin-positive B-lymphocytes, as well as higher average mean intensity of fluorescence, in the AD population (Figure 3). Note also that cells from the control population showed greater heterogeneity compared to AD patients by both measures. Variance in the controls is predicted from a population whose cognitive function levels are heterogeneous below the threshold of cognitive impairment. In contrast, the AD population has an extremely low variance, also as expected, because their cognitive function is compromised and they have crossed the “filipin threshold”. These differences in variance are consistent with the notion that MCI patients might be separated out in terms of their risk of conversion to AD, strongly supporting a potential clinical application for filipin testing. It is also noteworthy that the choice of B-lymphocytes was determined by their commercial availability. Further work in larger clinical studies will determine differences in filipin sensitivity between these cells and different subpopulations of PBMCs, which may show comparable or superior clinical value than B-lymphocytes to diagnose cognitive status and/or identify at-risk MCI populations. In summary, we conclude that quantifying cellular filipin levels can accurately differentiate between control and AD patients in blood cells.Our findings may help define AD in the blood by focusing on markers of cellular pathology, and they provide strong preliminary data in support of larger clinical studies to test whether afilipin-based method can indeed predict the risk of AD conversion in MCI patients. Acknowledgements The authors acknowledge Dr. Kimberly Payne for help with the isolation of PBMCs and the use of flow cytometry. References [1] Alzheimer’s Association (2013) 2013 Alzheimer’s Disease Facts and Figures. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association, 9, 208-245. http://dx.doi.org/10.1016/j.jalz.2013.02.003 [2] Golde, T.E., Petrucelli, L. and Lewis, J. (2010) Targeting Aβ and Tau in Alzheimer’s Disease, an Early Interim Report. Experimental Neurology, 223, 252-266. http://dx.doi.org/10.1016/j.expneurol.2009.07.035 [3] Castellani, R.J. and Perry, G. (2012) Pathogenesis and Disease-Modifying Therapy in Alzheimer’s Disease: The Flat Line of Progress. Archives of Medical Research, 43, 694-698. http://dx.doi.org/10.1016/j.arcmed.2012.09.009 [4] Castello, M.A. and Soriano, S. (2014) On the Origin of Alzheimer’s Disease. Trials and Tribulations of the Amyloid Hypothesis. Ageing Research Reviews, 13C, 10-12. http://dx.doi.org/10.1016/j.arr.2013.10.001 [5] Castello, M.A. and Soriano, S. (2013) Rational Heterodoxy: Cholesterol Reformation of the Amyloid Doctrine. Ageing Research Reviews, 12, 282-288. http://dx.doi.org/10.1016/j.arr.2012.06.007 [6] Rodríguez-Rodríguez, E., Mateo, I., Infante, J., Llorca, J., García-Gorostiaga, I., Vázquez-Higuera, J.L. et al. (2009) Interaction between HMGCR and ABCA1 Cholesterol-Related Genes Modulates Alzheimer’s Disease Risk. Brain Research, 1280, 166-171. http://dx.doi.org/10.1016/j.brainres.2009.05.019 [7] Jones, L., Holmans, P.A., Hamshere, M.L., Harold, D., Moskvina, V., Ivanov, D., et al. (2010) Genetic Evidence Implicates the Immune System and Cholesterol Metabolism in the Aetiology of Alzheimer’s Disease. PLoS ONE, 5, Article ID: e13950. http://dx.doi.org/10.1371/journal.pone.0013950 [8] Liu, J.-P., Tang, Y., Zhou, S., Toh, B.H., McLean, C. and Li, H. (2010) Cholesterol Involvement in the Pathogenesis of Neurodegenerative Diseases. Molecular and Cellular Neuroscience, 43, 33-42. http://dx.doi.org/10.1016/j.mcn.2009.07.013 [9] Naj, A.C., Jun, G., Beecham, G.W., Wang, L.-S., Vardarajan, B.N., Buros, J., et al. (2011) Common Variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 Are Associated with Late-Onset Alzheimer’s Disease. Nature Genetics, 43, 436-441. http://dx.doi.org/10.1038/ng.801 [10] Björkhem, I. (2012) Five Decades with Oxysterols. Biochimie, 95, 448-454. M. A. Castello et al. 143 http://dx.doi.org/10.1016/j.biochi.2012.02.029 [11] Pani, A., Dessì, S., Diaz, G., La Colla, P., Abete, C., Mulas, C., et al. (2009) Altered Cholesterol Ester Cycle in Skin Fibroblasts from Patients with Alzheimer’s Disease. Journal of Alzheimer’s Disease: JAD, 18, 829-841. [12] Pani, A., Mandas, A., Diaz, G., Abete, C., Cocco, P.L., Angius, F., et al. (2009) Accumulation of Neutral Lipids in Peripheral Blood Mononuclear Cells as a Distinctive Trait of Alzheimer Patients and Asymptomatic Subjects at Risk of Disease. BMC Medicine, 7, 66. http://dx.doi.org/10.1186/1741-7015-7-66 [13] Ghribi, O., Larsen, B., Schrag, M. and Herman, M.M. (2006) High Cholesterol Content in Neurons Increases BACE, β-Amyloid, and Phosphorylated Tau Levels in Rabbit Hippocampus. Experimental Neurology, 200, 460-467. http://dx.doi.org/10.1016/j.expneurol.2006.03.019 [14] Prasanthi, J.R.P., Feist, G., Thomasson, S., Thompson, A., Schommer, E. and Ghribi, O. (2008) Differential Effects of 24-Hydroxycholesterol and 27-Hydroxycholesterol on Tyrosine Hydroxylase and α-Synuclein in Human Neuroblastoma SH-SY5Y Cells. Journal of Neurochemistry, 107, 1722-1729. http://dx.doi.org/10.1111/j.1471-4159.2008.05736.x [15] Prasanthi, J.R.P., Huls, A., Thomasson, S., Thompson, A., Schommer, E. and Ghribi, O. (2009) Differential Effects of 24-hydroxycholesterol and 27-hydroxycholesterol on β-Amyloid Precursor Protein Levels and Processing in Human Neuroblastoma SH-SY5Y Cells. Molecular Neurodegeneration, 4, 1. http://dx.doi.org/10.1186/1750-1326-4-1 [16] Burg, V.K., Grimm, H.S., Rothhaar, T.L., Grösgen, S., Hundsdörfer, B., Haupenthal, V.J., et al. (2013) Plant Sterols the Better Cholesterol in Alzheimer’s Disease? A Mechanistical Study. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 33, 16072-16087. http://dx.doi.org/10.1523/JNEUROSCI.1506-13.2013 [17] Argoff, C.E., Kaneski, C.R., Blanchette-Mackie, E.J., Comly, M., Dwyer, N.K., Brown, A., Brady, R.O. and Pentchev, P.G. (1990) Type C Niemann-Pick Disease: Documentation of Abnormal LDL Processing in Lymphocytes. Biochemical and Biophysical Research Communications, 171, 38-45. http://dx.doi.org/10.1016/0006-291X(90)91353-T [18] Distl, R., Treiber-Held, S., Albert, F., Meske, V., Harzer, K. and Ohm, T.G. (2003) Cholesterol Storage and Tau Pathology in Niemann-Pick Type C Disease in the Brain. The Journal of Pathology, 200, 104-111. http://dx.doi.org/10.1002/path.1320 [19] Tamul, K.R., Schmitz, J.L., Kane, K. and Folds, J.D. (1995) Comparison of the Effects of Ficoll-Hypaque Separation and Whole Blood Lysis on Results of Immunophenotypic Analysis of Blood and Bone Marrow Samples from Patients with Hematologic Malignancies. Clinical and Diagnostic Laboratory Immunology, 2, 337-342. [20] Koh, C.H.V. and Cheung, N.S. (2006) Cellular Mechanism of U18666A-Mediated Apoptosis in Cultured Murine Cortical Neurons: Bridging Niemann-Pick Disease Type C and Alzheimer’s Disease. Cellular Signalling, 18, 1844-1853. http://dx.doi.org/10.1016/j.cellsig.2006.04.006 [21] Cenedella, R.J. (2009) Cholesterol Synthesis Inhibitor U18666A and the Role of Sterol Metabolism and Trafficking in Numerous Pathophysiological Processes. Lipids, 44, 477-487. http://dx.doi.org/10.1007/s11745-009-3305-7 [22] Arthur, J.R., Heinecke, K.A. and Seyfried, T.N. (2011) Filipin Recognizes both GM1 and Cholesterol in GM1 Gangliosidosis Mouse brain. Journal of Lipid Research, 52, 1345-1351. http://dx.doi.org/10.1194/jlr.M012633 [23] Mandas, A., Abete, C., Putzu, P.F., la Colla, P., Dessì, S. and Pani, A. (2012) Changes in Cholesterol MetabolismRelated Gene Expression in Peripheral Blood Mononuclear Cells from Alzheimer Patients. Lipids in Health and Disease, 11, 39. http://dx.doi.org/10.1186/1476-511X-11-39